

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://www.jocmr.org |
Review
Volume 14, Number 1, January 2022, pages 22-27

Experimental Cardiorenal Syndrome Type 3: What Is Known so Far?
Daniel Patschana, b, Benedikt Marahrensa, Monique Janscha, Susann Patschana, Oliver Rittera
aUniversitatsklinikum Brandenburg, Zentrum fur Innere Medizin I - Kardiologie, Angiologie und Nephrologie, Medizinische Hochschule Brandenburg, Brandenburg, Germany
bCorresponding Author: Daniel Patschan, Zentrum Innere Medizin 1, Universitatsklinikum Brandenburg, Medizinische Hochschule Brandenburg Hochstrabe 29, 14770 Brandenburg, Germany
Manuscript submitted November 19, 2021, accepted January 13, 2022, published online January 29, 2022
Short title: Experimental CRS Type 3
doi: https://doi.org/10.14740/jocmr4639
| Abstract | ▴Top |
Background: The concept of cardiorenal syndrome (CRS) has been established more than 10 years ago. Five distinct types of CRS have been defined. In CRS type 3, acute kidney injury (AKI) induces cardiac complications such as ventricular decompensation due to arrhythmias, myocardial ischemia, or fluid retention with or without arterial hypertension. The risk of cardiovascular events in AKI has been known for many years, even long before the introduction of the CRS concept. However, epidemiological and clinical studies published in recent years increasingly emphasized CRS type 3 (and the remaining four types also) as separate entity which requires particular therapeutic attention in an interdisciplinary manner. However, only a limited number of experimental studies specifically addressed CRS type 3 so far. Our review aims to summarize experimental studies on the pathological mechanisms in CRS type 3.
Methods: The following search criteria were employed in order to identify articles published on the topic: “cardiorenal syndrome 3” OR “cardiorenal syndrome type 3” OR “CRS type 3” OR “CRS 3” AND “experimental” OR “mouse” OR “mice” OR “rats” OR “animals”; additional criteria were “myocardium” AND “ischemia” AND “kidney” OR “renal”. By applying the search criteria mentioned earlier, 10 references were finally selected.
Results: By applying the search strategy, 10 experimental studies were finally selected. All included cardiac outcome analysis in AKI animals. The data clearly provide evidence for cardiac complications that evolve independently from excretory kidney dysfunction. Pathological processes that emerge in the heart of animals subjected to renal ischemia involve inflammation, a dysbalance of redox components, pro-apoptotic processes, and mitochondrial dysfunction.
Conclusion: The findings may explain why AKI increases the risk of acute cardiac complications even if dialysis treatment has been initiated.
Keywords: CRS type 3; AKI; Inflammation; Apoptosis; Mitochondrial dysfunction; Redox components
| Introduction | ▴Top |
Cardiorenal syndrome (CRS)
CRS, which has initially been described by Ronco and colleagues in 2008 [1], is increasingly being recognized as disorders with common pathophysiological characteristics. The concept “cardiorenal” emphasizes the interdisciplinary character of the diseases, not only from a pathophysiological but also a therapeutic perspective. Five distinct types are differentiated, and in all of these, heart and kidney function are variably impaired at the same time, either with acute or chronic onset: acute heart failure induces acute kidney injury (AKI) - CRS type 1; chronic heart failure accelerates progression of chronic kidney disease (CKD) - CRS type 2; AKI is followed by acute abnormalities in cardiac function - CRS type 3; CKD induces/aggravates chronic heart disease including cardiac hypertrophy - CRS type 4. In type 5 CRS finally, a systemic disorder such as sepsis or an autoimmune-mediated disease (e.g., systemic lupus erythematodes) affects both heart and kidneys. Table 1 summarizes characteristics, pathophysiological hallmarks, and treatment strategies of CRS types 1-4. The general idea behind the CRS concept was and is to provide a more standardized rationale for treating patients and to facilitate a more accurate design of clinical studies related to the topic. Finally, and this aspect has been emphasized in the literature since 2008, different types of CRS emerge from common pathophysiological processes. The latter involve numerous events including hemodynamic, metabolic, inflammatory and genetic factors.
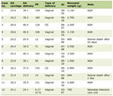 Click to view | Table 1. Characteristics, Pathophysiological Hallmarks, and Treatment Strategies of CRS Types 1-4 |
CRS type 3
AKI affects an increasing number of patients treated in hospitals in central Europe and the USA. It is being estimated that AKI establishes in up to 30% of all in-hospital patients [2]. The following criteria are currently used for AKI diagnosis: increase of serum creatinine of at least 0.3 mg/dL within 48 h, or a 1.5-fold increase within 7 days, or a reduction in urinary output to below 0.5 mL/kg/h for at least 6 h [3]. Early AKI diagnosis remains difficult, although substantial progress has been achieved with the identification of new biomarkers [4]. Particularly markers of structural kidney damage will most likely be incorporated into new diagnostic criteria or definitions of AKI [5]. The prognosis of AKI subjects has only mildly been improved over the years, since almost no therapeutic measures substantially promote renal recovery specifically, once AKI has been established. The term “acute cardiac dysfunction” must be used in a broader sense. It encompasses acute failure with low cardiac output, acute myocardial ischemia with or without ventricular failure, and arrhythmias [1]. Epidemiological data on CRS type 3 are limited. According to the definition, any cardiac complication due to AKI justifies the diagnosis. A retrospective investigation in 129 intensive care unit (ICU) subjects [6] performed between January 2006 and January 2008 identified an AKI incidence of 40%, and cardiac arrest was responsible for death in 20.3%. However, the actual prevalence of AKI-associated cardiac complications is higher without doubt since other consequences than cardiac arrest must be considered also: pulmonary congestion, arrhythmias of various etiology, pericarditis, and coronary artery insufficiency. Before the concept of CRS arose, the retention of water and solutes, followed by volume expansion, electrolyte and acid-base disorders such as hyperkalemia and acidosis have been proposed as the most important mechanisms responsible for cardiac complications in AKI. Meanwhile, the knowledge has significantly been expanded. On one hand, AKI induces renin-angiotensinogen-aldosterone system (RAAS) and sympathetic nervous system activation. It also activates the immune system in most situations, no matter what type of disease/condition was the primary cause [7]. Immune activation occurs in a systemic manner, accompanied by lymphocyte/monocyte and endothelial cell activation and numerous other events. Mulay and colleagues [8] provided an excellent summary of the topic. The inflammatory response in AKI may not be underestimated. On the contrary, AKI is for sure a systemic inflammatory disease [9].
Bagshaw et al [10] and Virzi and colleagues [11] published detailed review articles on current pathophysiological ideas in CRS type(s) 3 and 1, including data from human analyzes. Aim of the current article in contrast is to exclusively focus on experimental studies in the field.
| Methods | ▴Top |
The following search criteria were employed in order to identify articles published on the topic: “cardiorenal syndrome 3” OR “cardiorenal syndrome type 3” OR “CRS type 3” OR “CRS 3” AND “experimental” OR “mouse” OR “mice” OR “rats” OR “animals”; additional criteria were “myocardium” AND “ischemia” AND “kidney” OR “renal”. By applying the search criteria mentioned earlier, 10 references were finally selected. Figure 1 summarizes the selection process.
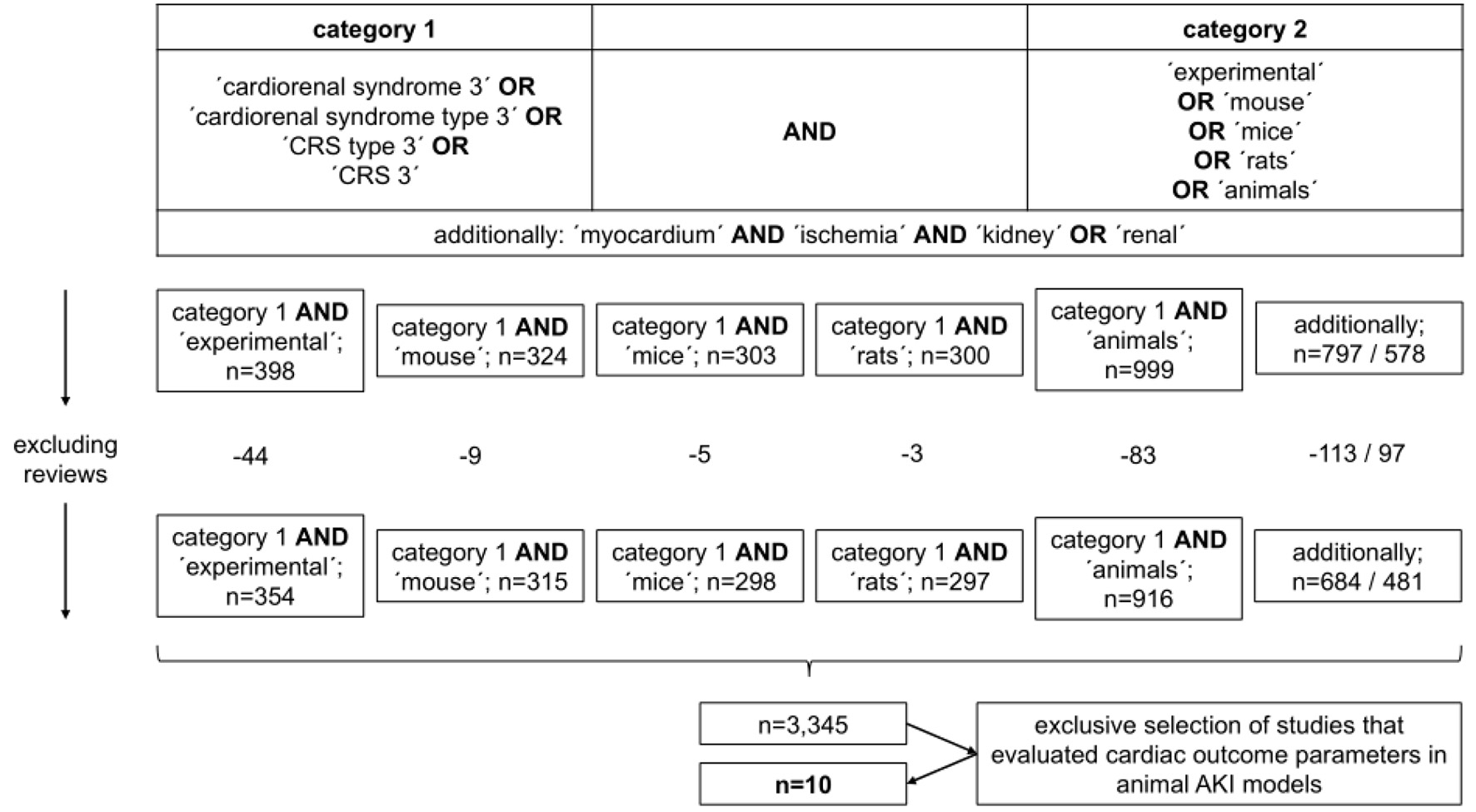 Click for large image | Figure 1. Selection process of references cited in the study. |
| Experimental Evidence in CRS Type 3 | ▴Top |
Mitochondrial reactions
More than 20 years ago, Robinson and colleagues [12] analyzed response patterns of cardiac preparations from rats with acute renal failure (ARF), induced by glycerol administration. The left atria showed inotropic incompetence in response to the calcium agonist Bay K 8644. Right ventricular myocardium however showed lower sensitivity to at least three inotropic mediators (isoprenaline, 3-isobutyl-1-methylxanthine, and Bay K 8644). Mitochondrial respiration was uncoupled, although not accompanied by alterations in the cellular adenine nucleotide or creatine phosphate contents or by impaired cellular energy charge. The pyruvate dehydrogenase activity, which indirectly reflects the mitochondrial calcium load, was significantly higher in isoprenaline treated hearts from ARF rats. Overall, the analyses showed impaired calcium homeostasis in ARF hearts, most likely responsible for diminished reactivity to inotropic substances.
Sumida et al [13] evaluated mitochondrial malfunction in murine hearts after renal ischemia reperfusion. Male C57BL6 mice were employed, and ischemia lasted for 30 min. At 24 h, cardiac mitochondria were significantly fragmented. At 72 h, cardiomyocytes displayed increased caspase-3 activity which indicated stimulation of apoptosis. At the same time, echocardiography revealed diminished fractional shortening of the left ventricle. Several proteins involved in regulating mitochondrial activity were quantified in both compartments, the mitochondria and the whole heart. Out of four candidates, only mitochondrial dynamin-related protein 1 (Drp1) increased post-ischemia. Also, cardiac tumor necrosis factor-alpha (TNF-α) was higher in post-ischemic animals. Finally, mitochondrial fragmentation was reduced after pre-treatment with mitochondrial division inhibitor-1 (mdivi-1), a substance with known inhibitory effects on Drp1.
Immune responses
In a 2003 published study, Kelly [14] performed renal artery clamping experiments in Sprague-Dawley rats, and ischemia lasted for 30 min. Systemic TNF-α levels transiently increased after ischemia, followed by normalization until 48 h. Myocardial TNF-α and interleukin-1 (IL-1) were subsequently elevated, beginning at 6 h until 48 h post-ischemia. In addition, cardiac intercellular cell adhesion molecule-1 (ICAM-1) mRNA abundances were elevated (6, 24, and 48 h). Extrarenal myeloperoxidase (MPO) activities, indicating leukocyte activation, increased in both organs, heart and liver. The latter effects were abrogated by the administration of anti-ICAM-1. Finally, ultrasound analysis showed impaired left ventricular function in post-ischemic rats. The study clearly showed that post-AKI systemic inflammation affects extrarenal tissues such as the heart in both, a functional and structural manner.
A critical role of interleukin-1beta (IL-1β) for the induction of cardiac arrhythmias post-AKI was shown by Alarcon and colleagues [15]. So-called NLrp3-/- and Casp1-/- mice were employed in renal ischemia reperfusion experiments, and both strains are characterized by the inability to produce and secrete IL-1β. Genetically modified animals did not suffer from ventricular arrhythmias at day 15 post-ischemia, as opposed to wild-type (WT) mice. Also, renal ischemia did neither induce renal insufficiency nor atrophy in the absence of IL-1β. Interestingly, macrophage depletion or anti-IL-1β treatment in WT mice had comparable effects on ventricular susceptibility to arrhythmias.
The effects of melatonin on cardiomyocyte function/metabolism were studied by Wang et al [16]. All experiments were performed in 8 weeks old, male C57BL6 mice. AKI was induced by bilateral renal ischemia for 30 min, and analyses were carried out 24 and 74 h later. Melatonin was applied intraperitoneally at 30 min after reperfusion. First of all, melatonin significantly protected post-ischemic animals from excretory kidney insufficiency. The hormone also prevented cardiac diastolic dysfunction. The latter effect went in line with diminished myocardial inflammation, as assessed by lower levels of the following proteins: TNF-α, IL-6, and transforming growth factor-beta (TGF-β). Cardiomyocyte viability was improved upon melatonin treatment as well. Two additional of myocardial parameters were stabilized: mitochondrial function and cytoplasmic calcium overload. Finally, the cardioprotective effects of melatonin were shown to depend on the so-called inositol 1, 4, 5-trisphosphate receptors-mitochondrial calcium uniporter (IP3R-MCU) signaling pathway.
Oxidative stress
Caio-Silva et al [17] performed unilateral ischemia reperfusion experiments in male C57BL/6 mice. At 8 and 15 days after an ischemic period of 60 min, kidneys and heart were semiquantitatively assessed for several oxidative/anti-oxidative stress parameters (redox balance components): catalase (CAT), superoxide dismutase (SOD), total antioxidant capacity (FRAP), NADPH oxidase (NOX), nitric oxide synthase (NOS), and hydrogen peroxide (H2O2). In addition, the authors evaluated tissue contents of NO donors such as S-nitrosothiol (RSNO). Intrarenal activities of antioxidant enzymes, such as SOD, were increased, and the NO bioavailability was higher as well. Myocardial analyses revealed elevated activities of both, NOX and NOS at day 8, combined with aggravated cell damage. On day 15 however, the amount of protein damage was reduced.
Others
A recent study by Wang and colleagues [18] aimed to identify the primary stimulus of myocardial damage in CRS type 3. It was emphasized that inflammation and oxidative stress indeed induce cardiac dysfunction. However, mediators capable to activate or initiate the complex processes involved in cardiac pathologies have been unknown so far. Again, male C57BL6 mice (8 weeks) were subjected to bilateral renal artery clamping for 30 min, and analyses were also performed 24 and 72 h later. Echocardiography did not reveal impaired systolic but diastolic function of the left ventricle in post-ischemic animals. The mitochondrial structure in cardiomyocytes was affected. Proteomic analyses from heart tissue showed an upregulation of more than 400 proteins, from which 91 candidates were involved in metabolic functions. So-called “STRING protein-protein interaction network analysis” allowed identifying growth factor receptor-bound protein-2 (Grb2) as a key regulator of AKI-associated cardiac pathology. Exogenous administration of a Grb2 inhibitor resulted in improved cardiac function, stabilization of cardiomyocyte metabolism, and reversal of mitochondrial malfunction. Finally, the Akt/mTOR pathway was identified as a mediator of Grb2. The authors also found elevated IL-6 levels post-AKI, and the cytokine unregulated Grb2 expression. Thus, the study revealed Grb2 as an inductor of cardiac damage in experimental CRS type 3.
A 2006 published experimental study was performed in male mongrel dogs [19]. Principal aim was to characterize coronary vasoregulation in the context of ischemia-induced ARF. Renal ischemia lasted for 30 min. The following parameters were analyzed: coronary pressure flow relations, distribution of myocardial blood flow, and coronary dose-responses to vasodilatory substances that either act in an endothelium-dependent or -independent manner. The latter series of experiments showed significantly impaired vascular reactivity to all of the applied compounds. Also, myocardial oxygen consumption was higher in ARF as compared to control dogs (resting conditions). Finally, ARF dogs displayed reduced coronary vascular conductance. In summary, the data indicated ARF-associated vascular dysfunction, most likely induced by a systemic dysbalance of vasomodulatory substances.
In 2019, Trentin-Sonoda and colleagues [20] analyzed the role of Casp1 in mediating the cardiac response to renal ischemia reperfusion injury. Particularly, the authors assessed cardiac hypertrophy and apoptosis. Male C57/BL/6 and Casp1 knockout mice were subjected to unilateral renal ischemia for 60 min, followed by a latency period of 15 days. The lack of Casp1 was associated with increased cardiac hypertrophy. Also, caspases 3, 7, and 9 activities were higher, indicating the stimulation of apoptosis in an IL-1β-independent manner.
A 1996 published study [21] evaluated the so-called area at risk (AR) and the infarction area (IA) in rats that underwent transient coronary artery occlusion for 60 min. Aim was to determine whether brief occlusion of either the mesenteric or the left renal artery may prevent myocardial damage. While renal ischemia failed to protect the heart under normothermic conditions, lowering the body temperature to 30-31°C was associated with significant cardioprotection of ischemic preconditioning (reduction of the IA/AR ratio).
Figure 2 summarizes pathophysiological aspects of CRS type 3 with regard to experimental evidence.
 Click for large image | Figure 2. Molecular mechanisms involved in myocardial dysfunction in CRS type 3. The figure summarizes the essential findings described in the article. Acute kidney injury of various origin triggers mitochondrial dysfunction in cardiomyocytes, an aberrant (systemic and local) immune response, and systemic vasomodulatory disturbances. CRS: cardiorenal syndrome. |
| Conclusions | ▴Top |
Experimental data support the concept of CRS type 3.
The studies presented herein show that cardiac pathologies in CRS type 3 evolve from pathophysiological events, which are heterogenous in terms of origin and dynamics. Intra-renal activation of the immune system and the production of vasoconstrictive substances may be proposed as key events. Both act as danger signals in a systemic manner.
Subsequently, cardiac mitochondriopathy, an impaired balance of redox components, and activation of the pro-apoptotic machinery induce myocardial damage. All processes occur in the absence of endogenous toxification or fluid retention due to diminished excretory kidney function.
This may partly explain the significantly higher cardiovascular risk of AKI subjects, even if renal replacement therapy has been initiated.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author Contributions
DP wrote the article. BM assisted in writing and prepared Figure 2. MJ assisted in the literature search. SP prepared Figure 1. OR designed the article and helped in writing. All authors approved the final version of the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
AKI: acute kidney injury; ARF: acute renal failure; Casp1: caspase 1; CAT: catalase; CKD: chronic kidney disease; CRS: cardiorenal syndrome; Drp1: dynamin-related protein 1; Grb2: growth factor receptor-bound protein-2; ICAM-1: intercellular cell adhesion molecule-1; IP3R-MCU: inositol 1, 4, 5-trisphosphate receptors-mitochondrial calcium uniporter; IL-1β: interleukin-1beta; IL-6: interleukin-6; MPO: myeloperoxidase; NLrp3: NOD-, LRR- and pyrin domain-containing protein 3; NADPH: nicotinamide adenine dinucleotide phosphate hydrogen; NOS: nitric oxide synthase; SOD: superoxide dismutase; TGF-β: transforming growth factor-beta; TNF-α: tumor necrosis factor-alpha; WT: wild type
| References | ▴Top |
- Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52(19):1527-1539.
doi pubmed - Hoste EAJ, Kellum JA, Selby NM, Zarbock A, Palevsky PM, Bagshaw SM, Goldstein SL, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol. 2018;14(10):607-625.
doi pubmed - Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120(4):c179-184.
doi pubmed - Schrezenmeier EV, Barasch J, Budde K, Westhoff T, Schmidt-Ott KM. Biomarkers in acute kidney injury - pathophysiological basis and clinical performance. Acta Physiol (Oxf). 2017;219(3):554-572.
doi pubmed - Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52.
doi pubmed - de Abreu KL, Silva Junior GB, Barreto AG, Melo FM, Oliveira BB, Mota RM, Rocha NA, et al. Acute kidney injury after trauma: Prevalence, clinical characteristics and RIFLE classification. Indian J Crit Care Med. 2010;14(3):121-128.
doi pubmed - Patschan D, Kribben A, Muller GA. Postischemic microvasculopathy and endothelial progenitor cell-based therapy in ischemic AKI: update and perspectives. Am J Physiol Renal Physiol. 2016;311(2):F382-394.
doi pubmed - Mulay SR, Holderied A, Kumar SV, Anders HJ. Targeting inflammation in so-called acute kidney injury. Semin Nephrol. 2016;36(1):17-30.
doi pubmed - Singbartl K, Formeck CL, Kellum JA. Kidney-immune system crosstalk in AKI. Semin Nephrol. 2019;39(1):96-106.
doi pubmed - Bagshaw SM, Hoste EA, Braam B, Briguori C, Kellum JA, McCullough PA, Ronco C. Cardiorenal syndrome type 3: pathophysiologic and epidemiologic considerations. Contrib Nephrol. 2013;182:137-157.
doi pubmed - Virzi G, Day S, de Cal M, Vescovo G, Ronco C. Heart-kidney crosstalk and role of humoral signaling in critical illness. Crit Care. 2014;18(1):201.
doi pubmed - Robinson SC, Bowmer CJ, Yates MS. Cardiac function in rats with acute renal failure. J Pharm Pharmacol. 1992;44(12):1007-1014.
doi pubmed - Sumida M, Doi K, Ogasawara E, Yamashita T, Hamasaki Y, Kariya T, Takimoto E, et al. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol. 2015;26(10):2378-2387.
doi pubmed - Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003;14(6):1549-1558.
doi pubmed - Alarcon MML, Trentin-Sonoda M, Panico K, Schleier Y, Duque T, Moreno-Loaiza O, de Yurre AR, et al. Cardiac arrhythmias after renal I/R depend on IL-1beta. J Mol Cell Cardiol. 2019;131:101-111.
doi pubmed - Wang J, Toan S, Li R, Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem Pharmacol. 2020;174:113832.
doi pubmed - Caio-Silva W, da Silva Dias D, Junho CVC, Panico K, Neres-Santos RS, Pelegrino MT, Pieretti JC, et al. Characterization of the oxidative stress in renal ischemia/reperfusion-induced cardiorenal syndrome type 3. Biomed Res Int. 2020;2020:1605358.
doi pubmed - Wang J, Sun X, Wang X, Cui S, Liu R, Liu J, Fu B, et al. Grb2 induces cardiorenal syndrome type 3: roles of IL-6, cardiomyocyte bioenergetics, and Akt/mTOR pathway. Front Cell Dev Biol. 2021;9:630412.
doi pubmed - Kingma JG, Jr., Vincent C, Rouleau JR, Kingma I. Influence of acute renal failure on coronary vasoregulation in dogs. J Am Soc Nephrol. 2006;17(5):1316-1324.
doi pubmed - Trentin-Sonoda M, Fratoni FM, da Cruz Junho CV, Silva WC, Panico K, Carneiro-Ramos MS. Caspase-1 as molecular key in cardiac remodeling during cardiorenal syndrome type 3 in the murine model. Curr Mol Med. 2019;20(1):72-78.
doi pubmed - Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD. Myocardial protection by brief ischemia in noncardiac tissue. Circulation. 1996;94(9):2193-2200.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.