

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 12, Number 5, May 2020, pages 286-292
The Issue of the Antiphospholipid Antibody Syndrome
Dinaldo C. Oliveiraa, b, Augusto Correiaa, Carolina Oliveiraa
aHospital das Clinicas, Federal University of Pernambuco, Recife-PE, Brazil
bCorresponding Author: Dinaldo C. Oliveira, Hospital das Clinicas, Federal University of Pernambuco, Rua Prof. Moraes Rego1235, CEP 50670-901, Recife-PE, Brazil
Manuscript submitted April 5, 2020, accepted April 15, 2020
Short title: Antiphospholipid Syndrome
doi: https://doi.org/10.14740/jocmr4154
- Abstract
- Introduction
- Epidemiology
- Pathophysiology
- Clinical Findings
- Diagnosis
- Management
- Prophylaxis
- Prognosis
- Conclusions
- References
| Abstract | ▴Top |
Antiphospholipid antibody syndrome (APS) is a state of hypercoagulability secondary to an autoimmune disorder. It is associated with thrombotic events in venous and arterial vessels, obstetric complications characterized by recurrent fetal losses, and increased perinatal morbidity. APS is classified as primary, when not associated with other pathologies; or secondary, when associated with an underlying autoimmune disease with, solid tumor, or hematological disorder. Clinical findings include livedo reticularis, thrombocytopenia or hemolytic anemia, maternal morbidity, and recurrent thrombotic episodes and others. Laboratory tests show circulating antiphospholipid antibodies (aPLs); however, even in the presence of these antibodies, patients can be asymptomatic. Estimates predict that about 5% of the populations have circulating aPLs, but the incidence of APS is only five cases per 100,000 people, as diagnosis of this syndrome requires clinical and laboratory findings to be simultaneously present. In cases of secondary APS, or in acute cases with imminent risk of death (as in catastrophic APS), it may be necessary to reduce aPL serum levels using immunomodulators, immunosuppressants, or plasmapheresis, in order to treat the associated pathologies. In other situations, the use of immunotherapy is not indicated. In other patients heparin, aspirin or anticoagulants either alone or associated should be administered depending on each specific case.
Keywords: Antiphospholipid antibody syndrome; Arterial thrombosis; Venous thrombosis; Repetitive abortion
| Introduction | ▴Top |
Antiphospholipid antibody syndrome (APS) is defined by the development of (often multiple) venous or arterial thromboses or by the occurrence of maternal morbidity, primarily recurrent fetal losses, in the presence of antiphospholipid antibodies (aPLs). These aPLs include lupus anticoagulant (LA), anticardiolipin antibodies (aCLs), or antibodies against β2 glycoprotein-1 (β2GPI) [1-8].
APLs are a heterogeneous group of autoantibodies clearly associated with the development of thrombosis, as well as increased maternal morbidity and mortality, which together constitute APS. The term “antiphospholipid antibody syndrome” has been used from the 1980s to describe this condition, which consists of a type of autoantibody-induced thrombophilia [7].
APS typically occurs in the fourth decade of life and is classified as primary when not associated with other pathologies, or secondary when associated with an underlying autoimmune disease, solid tumor, or hematological disorder. Approximately 10-40% of patients with systemic lupus erythematosus (SLE), and up to 20% of patients with rheumatoid arthritis, are aPL positive. SLE patients who are LA positive are more likely to develop thrombotic events [9].
Thrombotic events can occur in any tissue or organ, and must be objectively confirmed by imaging or histopathology; there should be no association between thrombosis and the presence of an inflammatory process in the vessel wall [7-9].
Although many APS patients have other associated autoimmune disorders, APS thrombotic events are not associated with histological evidence of vasculitis, although inflammatory mediators and the activation of endothelial cells, monocytes and neutrophils are associated with the disease pathogenesis [7].
APS can eventually affect any organ, since there is a close relationship between the development of the disease and endothelial dysfunction. Increasing evidence shows that binding of aPLs to membrane receptors on endothelial cells results in an increased risk of thrombosis, accelerated atherosclerosis, acute myocardial infarction (AMI), and cerebrovascular accidents in APS patients. In addition, APS has been associated not only with adverse cardiovascular events but also with subclinical markers associated with endothelial dysfunction, such as increased intima-media thickness of the carotid arteries and changes in the ankle-brachial index [1-7].
Despite of significant advances in identifying the pathogenic characteristics of the disease, the pathophysiology of these complications is still not well understood, and the heterogeneity of APS suggests that more than one pathogenic process may be involved [1, 7, 8].
The main objective of this review is to emphasize to physicians that APS may be the cause of several common clinical presentations in medical practice; and it is important that this syndrome be considered and investigated (by physicians and not only by specialists) depending on patient’s characteristics; because if this syndrome is forgotten and untreated, it may have a negative impact on the prognosis.
| Epidemiology | ▴Top |
The prevalence of aPLs in the general population is estimated to be between 1% and 5% [8]. However, the presence of these antibodies can correlate with different scenarios: 1) Asymptomatic aPL carriers; 2) Patients with non-thrombotic clinical signs, such as thrombocytopenia, hemolytic anemia, or livedo reticularis; 3) Patients with venous or arterial thrombotic events; 4)Healthy women with a history of maternal morbidity.
The APS diagnosis is confirmed in only a minority of these patients [1-5]. Some studies indicate that the incidence of the syndrome is approximately five new cases per 100,000 people per year, with a prevalence of approximately 40 to 50 cases per 100,000 people. Catastrophic APS has a low prevalence in these patients, corresponding to less than 1% of all cases [10].
APS typically occurs in the fourth decade of life, and women are predominantly affected, especially in cases of secondary disease. Furthermore, there is no influence of race in APS [1].
| Pathophysiology | ▴Top |
Regardless of significant advances in identifying the pathogenic characteristics of APS, the pathophysiology of these complications is still not well understood. For more than a decade, it has been suggested that endothelial cells play a central role in the origin of the disease and may represent the common pathway through which autoimmunity and inflammation occurs [11].
Although circulating aPLs and the underlying endothelial dysfunction are the first triggers, a sustained inflammatory stimulus is needed to cause a thrombotic event. Infections, oxidative stress, and significant physical stress such as major surgery, can result in disease exacerbation [7].
Endothelial cell dysfunction mediated by the binding of aPLs to their β2GPI receptors, results in an increased risk of thrombosis, accelerated atherosclerosis, AMI and cerebrovascular accidents. In this regard a recent study associated maternal morbidity in APS with placental inflammatory responses mediated by the binding of aPLs to β2GPI receptors on the surface of placental trophoblasts [12].
APS patients have lower levels of nitric oxide (NO) in the endothelium, as there is a reduction in the activity of the endothelial nitric oxide synthase (eNOS) enzyme. The production of NO is important to maintain normal endothelial function and vascular health [1, 7].
Experimental models showed that aPL-mediated eNOS inhibition is the molecular basis of endothelial dysfunction, increased leukocyte cell adhesion to the endothelium, and thrombus formation. The binding of circulating aPLs to domain I of β2GPI receptors on endothelial cells induces dimerization. The subsequent interaction between the dimerized β2GPI and the apolipoprotein E receptor 2 mediates aPL-induced eNOS inhibition, increases endothelial adhesion molecule expression, and increases endothelin-1 and tissue factor production, leading to thrombus formation [13].
The binding of circulating aPLs to domain I of β2GPI receptors on platelets also induces thrombotic events. This is due to increased thromboxane production. Production of thromboxane A2, a potent platelet activator, leads to greater platelet adhesion to endothelial collagen and a subsequent increase in platelet aggregation [1].
In addition to eNOS and platelet aggregation inhibition, the thrombogenic effects of aPLs also involve activation of the classical pathway of the complement system. Complement activation and deposition were associated with placental lesions in female APS patients with obstetric complications. Some studies suggest that heparin prevents obstetric complications by blocking the activation of the complement system rather than directly preventing placental thrombosis [14].
Endothelial dysfunction mediated by circulating aPLs is associated with accelerated peripheral and coronary atherosclerosis in comparison to general population with identical risk factors. The premature onset of atherosclerosis in these patients is related to immunomodulatory, pro-inflammatory, and risk factors [13]. The aggressive control of classic cardiovascular risk factors is recommended in this group of patients [1, 7, 13]. Figure 1 shows a summary of the pathophysiology.
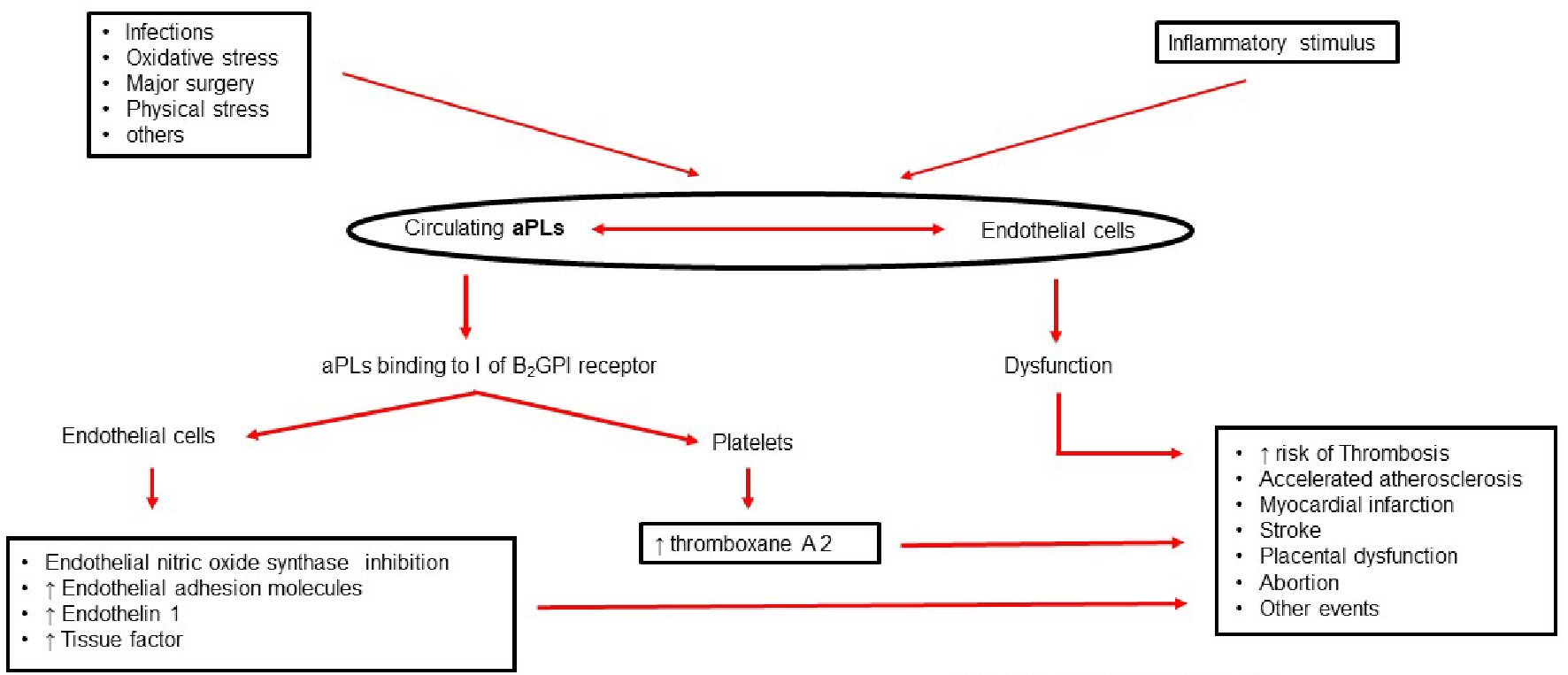 Click for large image | Figure 1. Antiphospholipid antibody syndrome: pathophysiology. aPLs: antiphospholipid antibodies. |
| Clinical Findings | ▴Top |
The clinical findings of APS are characterized by venous and arterial thrombosis, maternal morbidity, and moderate thrombocytopenia. Ranging from single vessel involvement to multiple vascular occlusions, it can result in a wide variety of clinical presentations [1-10].
Any combination of vascular occlusive events can occur in the same patient, with the time interval between them also varying considerably, from weeks to months or even years. Thus, clinical signs represent the direct or indirect consequences of the hypercoagulability state and associated thrombosis [8].
Maternal morbidities in APS are secondary to placental vascular bed changes that lead to the development of vascular insufficiency and structural damages. In this context, the most common obstetric signs are early (< 10 weeks) and late (≥ 10 weeks), miscarriages, followed by premature labor, preeclampsia, and intrauterine growth restriction (IUGR) [10].
The clinical presentation of APS is quite variable, with the spectrum of findings ranging from the presence of livedo reticularis and thrombocytopenia to the development of hemolytic anemia and recurrent thrombotic episodes. The most severe form of the syndrome is called catastrophic APS, which although rare, has a mortality rate greater than 50%. Catastrophic APS is characterized by the presence of microthrombi in several different vascular beds (at least three in an interval of less than 7 days) that result in multiple organ and system dysfunction, are associated with positive laboratory evidence of aPLs, and have no other alternative diagnosis [10].
A prospective cohort study evaluated 1,000 APS patients over a 10-year period and identified the most frequent clinical events in these patients. Deep vein thrombosis (DVT) was the most common clinical sign, followed by thrombocytopenia, livedo reticularis, cerebrovascular accident, pulmonary embolism, and AMI. The study reported thrombotic events in 16.6% of the patients during the first 5-year period and in 14.4% during the second 5-year period, and reported that 90.7% of the patients were alive after 10 years. Despite of the high survival rates, these data reflect a high morbidity in APS patients [10].
Table 1 shows the most frequent clinical manifestations of antiphospholipid antibodies syndrome.
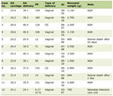 Click to view | Table 1. Possible Clinical Manifestations of Antiphospholipid Syndrome |
In addition to the presence of circulating aPLs, laboratory tests can detect changes suggesting direct Coombs-positive hemolytic anemia, and the presence of thrombocytopenia by a slight extension during coagulation tests [8].
| Diagnosis | ▴Top |
A preliminary set of diagnostic criteria for APS was established after a convention held in Sapporo, Japan, in 1999. In 2006, another convention was held in Sydney, Australia, in which a group of specialists proposed changes to the previous criteria, including the addition of anti-β2GPI antibody testing; these are currently the criteria used to define APS diagnosis [1, 7, 8].
The definitive diagnosis of this syndrome is established by the presence of at least one clinical criterion and one laboratory criterion (Table 2) [10].
Click to view | Table 2. Laboratory Criteria for the Diagnosis of Antiphospholipid Syndrome |
APS should be considered in cases of thrombosis in patients under 55 years of age. Risk-stratification of the etiological causes of thrombosis should be analyzed in cases with coexisting additional risks for thrombosis, such as age (> 55 years in men and > 65 years in women), or the presence of risk factors for cardiovascular diseases, such as systemic arterial hypertension, diabetes mellitus, dyslipidemia, smoking, hereditary thrombophilia, use of oral contraceptives, neoplasms, prolonged immobilization, and a history of recent surgery [1].
Although APS is an acquired immune-mediated thrombophilia, which is often associated with other autoimmune pathologies, its thrombotic events do not occur in combination with the vasculitis process. For this reason, thrombosis must be confirmed by imaging tests, Doppler ultrasound or histopathological analysis, except in cases of superficial venous thrombosis [7].
Concerning maternal morbidity, fetal losses should be documented by ultrasound or direct examination of the fetus. Fetal surveillance tests should be used in the evaluation of placental insufficiency, such as the detection of abnormal waveforms in flow velocity using Doppler ultrasound (suggesting fetal hypoxemia), the presence of oligohydramnios, or a postnatal weight lower than the 10th percentile for gestational age [1].
Although some clinical findings have not been included in the diagnostic criteria for APS, such as cardiac valve involvement, livedo reticularis, thrombocytopenia, and cognitive dysfunction, among other criteria described in Table 1, they are also associated with this syndrome. However, the current APS diagnostic criteria provide a more uniform basis, by which to select patients in whom this syndrome should be investigated, since the patient’s individual risk-stratification is emphasized [15].
One study showed that the current APS diagnostic criteria have a sensitivity of 90.3%, a specificity of 99.4%, a positive predictive value of 99.4%, and a negative predictive value of 91.1%. Thus, the use of these criteria limits the heterogeneity of patient groups for APS diagnosis, reducing the costs of complementary diagnostic evaluations and allowing more rapid determination of the correct etiological diagnosis [10, 15].
At the end of this disease spectrum is the most severe form of APS, catastrophic APS. Despite being rare, it has a mortality rate greater than 50%, and is characterized by the presence of microthrombi in several different vascular beds (at least three in an interval of less than 7 days), which lead to multiple organ and system dysfunction, and are associated with aPL-positive laboratory tests and no other alternative diagnosis [10, 16].
Vessel occlusions are generally also confirmed by imaging techniques. Renal involvement is defined by a 50% increase in serum creatinine, severe systemic arterial hypertension and/or proteinuria (> 500 mg in 24 h). If the patient was not previously diagnosed with APS, laboratory confirmation requires antiphospholipid antibody testing on two or more occasions at least 6 weeks apart [10].
| Management | ▴Top |
In cases of secondary APS, or in acute cases with imminent risk of death (as in catastrophic APS), it may be necessary to reduce aPL serum levels using immunomodulation therapies, e.g., high-dose corticosteroids, immunosuppressants, or plasmapheresis, in order to treat the associated pathologies. In other situations, the use of immunotherapy is not indicated, as the resulting reduction in circulating aPL levels is temporary and the antibodies quickly return to baseline levels after the therapy is discontinued. In cases of patients who have already developed thrombotic events, lifelong anticoagulation is an essential part of treatment [8].
Patients with APS defined by a first event of venous thrombosis should receive oral anticoagulant therapy with a target international normalized ratio (INR) between 2 and 3. In cases of arterial thrombosis, there is still controversy in the literature as to whether patients should receive oral anticoagulant therapy with a target INR around 3, or a combined anticoagulant and antiplatelet therapy with a target INR between 2 and 3; both approaches are accepted [16].
Although several new therapies have been developed in recent years, the treatment of choice is still vitamin K antagonist (VKA), and anticoagulant agent, such as warfarin [1, 8, 16].
Thrombocytopenia may occur during APS treatment, which is generally mild and does not require any active intervention. However, it can be severe in a minority of cases, requiring therapeutic interventions, such as the use of corticosteroids, and in some extreme cases, the use of immunomodulatory therapy with agents, such as azathioprine, intravenous immunoglobulins or rituximab [1, 8].
In cases of catastrophic APS, the treatment is aggressive, combining anticoagulation with heparin, high-dose steroids, plasmapheresis, and/or intravenous immunoglobulin infusion. Patients with refractory catastrophic APS may require the alternative use of immunobiological agents, such as rituximab and eculizumab [17].
The effectiveness of direct oral anticoagulants (DOACs) such as dabigatran, rivaroxaban, apixaban, etc., remains unclear. Some studies showed that approximately 20% of APS patients undergoing DOAC treatment presented vascular events during a mean follow-up period of 12 months, while one-third of the patients presented recurrent events during a 2-year follow-up [18]. The use of DOAC in APS patients is currently recommended only in cases of warfarin allergy or intolerance, or with poor anticoagulation control with this medication [16].
The TRAPS study was an investigator-sponsored, randomised, open-label, multicenter study with blinded endpoint adjudication (clinicaltrials.gov NCT02157272). Outcomes with rivaroxaban were compared with warfarin in patients with antiphospholipid syndrome and a history of thrombosis, and at high risk for thromboembolic events (patients who persistently tested positive for all three antiphospholipid tests) [19].
The trial was terminated prematurely after the enrolment of 120 patients due to an excess of thromboembolic events among patients in the rivaroxaban arm. Mean follow-up was 569 days. In the study, 59 patients were randomly assigned to rivaroxaban 20 mg (15 mg dose for patients with creatinine clearance < 50 mL/min) and 61 to warfarin (INR 2.0 - 3.0) [19].
Thromboembolic events occurred in 12% of patients assigned to receive rivaroxaban (four cases of ischemic stroke and three of myocardial infarction). No thromboembolic events were reported in patients assigned to receive warfarin. Major bleeding events occurred in four patients (7%) in the rivaroxaban group, and two patients (3%) in the warfarin group. No deaths were reported [19].
Available data for apixaban, edoxaban and dabigatran etexilate are more limited than for rivaroxaban because there have been no completed clinical trials of these products in patients with antiphospholipid syndrome. However, available data suggest these other DOACs may be associated with a similarly increased risk of recurrent thrombotic events as with the use of rivaroxaban [19].
One investigator-sponsored research study is ongoing to study rates of thrombosis in patients with antiphospholipid syndrome on apixaban (ASTRO-APS; Clinicaltrials.gov identifier NCT02295475). The final results are not yet available [19].
In summary, DOACs are not recommended in patients with antiphospholipid syndrome, particularly high-risk patients; it is advised to review whether continued treatment with a DOAC is appropriate for patients diagnosed with antiphospholipid syndrome, particularly high-risk patients, and consider switching to a vitamin K antagonist such as warfarin [19].
The evidence for the use of other anticoagulant agents in APS, such as fondaparinux or argatroban, is also limited; their use is recommended in cases of heparin-induced thrombocytopenia [1, 20].
As for the use of statins in APS patients, hitherto there are no formal recommendations for their use in patients with a normal lipid profile. However, statins can be considered in patients with refractory APS in spite of adequate anticoagulation, since these medications have an anti-inflammatory action on the vascular endothelium [1, 16].
Rituximab, an anti-CD20 (cluster of differentiation 20) monoclonal antibody, has been effectively used in cases of catastrophic APS, anticoagulant failure, recurrent thrombosis, and thrombocytopenia. A retrospective single-center study reported that rituximab decreased thrombotic events in APS patients with associated SLE and recurrent thrombosis despite adequate warfarin therapy. It is important to note that immunosuppression was added to the ongoing anticoagulant treatment [1, 17].
The current recommendations published in the ninth edition of the American College of Chest Physicians Guidelines propose the treatment of obstetric APS with a combination of either unfractionated or low-molecular-weight heparin in combination with low dose of aspirin (75 to 100 mg/day) [21].
In cases of patients already using warfarin, the therapy should be replaced by heparin in combination with aspirin. This transition should start immediately after pregnancy confirmation to avoid teratogenicity. The risk of warfarin-induced effects on the fetus is greater during the first 6 weeks of pregnancy, especially for patients using more than 5 mg per day [1, 21]. In cases of high-risk patients, when important risk factors persist after delivery, the guidelines suggest prolonged prophylaxis for up to 6 weeks after delivery [20].
Anticoagulant agents should be continued indefinitely for APS patients with a previous history of thrombotic disease [21].
The clinical manifestations described due to APS may occur as a result of other diseases. A detailed medical history and physical examination help physicians to think about possible causes of these clinical manifestations. It is important to have in mind that APS is one possibility to be thought.
The patient with DVT due to APS must be treated with VKA. Unlike other causes in which DOAC is an option, patient who had an acute or chronic coronary syndrome and underwent percutaneous coronary intervention should take dual antiplatelet therapy; but if this patient has APS, firstly he should take triple therapy, then VKA and P2Y12 receptor inhibitor for a while, and later only CKA. Patient who had ischemic stroke not due to APS should take aspirin; but if APS is related, he must receive VKA. For treatment of pulmonary embolism DOAC are an option, but not in those with APS.
Therefore, when one patient has one of the clinical manifestations described in Table 1, the search for a diagnosis of APS is important because it requires treatment with VKA, which may not happen with the most of the other causes. It is clear if the diagnosis is not given to those who have APS, the treatment will not be appropriate, so there will be risks to the patients
Once the diagnosis of APS is confirmed, the patient should be referred to a specialist.
| Prophylaxis | ▴Top |
APS patients who have never had a thrombotic event should be strictly managed for common cardiovascular risk factors as part of primary APS prophylaxis. Systemic arterial hypertension, dyslipidemia, and central obesity have been associated with thrombosis in aPL-positive patients, as these all increase damage to the vascular endothelium. Currently, all aPL-positive patients should receive appropriate thromboprophylaxis with heparin in high-risk situations, such as surgery, the postpartum period, and during prolonged immobilization [1, 16].
In cases of aPL-positive pregnant patients, the use of low-dose aspirin (50 - 100 mg per day) is recommended from the beginning of pregnancy until delivery to prevent fetal loss. Additionally, strict monitoring of the pregnancy is recommended to allow the early detection of placental vascular insufficiency signs [1, 8].
Hydroxychloroquine in combination with low-dose aspirin is recommended only for aPL-positive patients with SLE. There are no recommendations on the use of this medication for the primary prevention of thrombotic events in other aPL-positive populations [16, 21].
| Prognosis | ▴Top |
At 10-year follow-up the majority of patients with APS were alive according to an important European study. However, the morbidity in these patients was high [10].
A study on pregnant APS women showed that patients who were triple positive for aPLs (LA, aCL and anti-β2GPI positive), and/or had a history of previous thrombosis, were more likely to develop maternal morbidity than those positive for one or two aPLs with no history of thrombosis [6].
Several studies have tried to identify the individual risk of thrombosis in aPL-positive patients. More recently, a global antiphospholipid syndrome score (GAPSS) was developed in a cohort of 211 SLE patients from a single center [22]. The GAPSS is based on the combination of the independent risks for thrombosis and pregnancy loss, considering a panel of seven different aPLs, conventional cardiovascular risk factors, and autoimmune profiles (i.e., presence of autoantibodies). Each variable corresponds to a score and a cutoff value ≥ 10 points seems to be associated with increased risks of thrombosis or maternal morbidity in the context of SLE. Although the GAPSS has not yet been validated by other groups, it may become a useful tool in APS risk-stratification [6, 22].
| Conclusions | ▴Top |
APS is a rare disease and its pathophysiology is not fully understood yet. The clinical findings of APS are characterized by venous and arterial thrombosis, maternal morbidity, and moderate thrombocytopenia. Ranging from single vessel involvement to multiple vascular occlusions, it can result in a wide variety of clinical presentations. The definitive diagnosis of this syndrome is established by the presence of at least one clinical criterion and one laboratory criterion. The treatment is based on anticoagulants, antiplatelet, immunomodulators, immunosuppressants, or plasmapheresis and should be tailored according to patient’s characteristics. Despite of the chance of survival at 10 years after the diagnosis being high, the morbidity in patients with APS is significant.
Acknowledgments
We would thank the CAS Aortic, from Zurich (inspiration).
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Author Contributions
Dinaldo Oliveira: acquisition and analysis of data, draft work, and final approval; Augusto Correia: acquisition and analysis of data, draft work, and final approval; Carolina Oliveira: acquisition and analysis of data.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Corban MT, Duarte-Garcia A, McBane RD, Matteson EL, Lerman LO, Lerman A. Antiphospholipid syndrome: role of vascular endothelial cells and implications for risk stratification and targeted therapeutics. J Am Coll Cardiol. 2017;69(18):2317-2330.
doi pubmed - Vreede AP, Bockenstedt PL, Knight JS. Antiphospholipid syndrome: an update for clinicians and scientists. Curr Opin Rheumatol. 2017;29(5):458-466.
doi pubmed - Nazir S, Tachamo N, Lohani S, Hingorani R, Poudel DR, Donato A. Acute myocardial infarction and antiphospholipid antibody syndrome: a systematic review. Coron Artery Dis. 2017;28(4):332-335.
doi pubmed - Ahluwalia J, Sreedharanunni S. The Laboratory Diagnosis of the Antiphospholipid Syndrome. Indian J Hematol Blood Transfus. 2017;33(1):8-14.
doi pubmed - Lim W. Antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program. 2013;2013:675-680.
doi pubmed - Mineo C. Inhibition of nitric oxide and antiphospholipid antibody-mediated thrombosis. Curr Rheumatol Rep. 2013;15(5):324.
doi pubmed - Arachchillage DRJ, Laffan M. Pathogenesis and management of antiphospholipid syndrome. Br J Haematol. 2017;178(2):181-195.
doi pubmed - Cervera R. Antiphospholipid syndrome. Thromb Res. 2017;151(Suppl 1):S43-S47.
doi - Petri M. Epidemiology of the antiphospholipid antibody syndrome. J Autoimmun. 2000;15(2):145-151.
doi pubmed - Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de Ramon E, Buonaiuto V, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. 2015;74(6):1011-1018.
doi pubmed - Blum A, Simsolo C. The antiphospholipid syndrome and endothelial function. Isr Med Assoc J. 2004;6(9):556-558.
- Mulla MJ, Brosens JJ, Chamley LW, Giles I, Pericleous C, Rahman A, Joyce SK, et al. Antiphospholipid antibodies induce a pro-inflammatory response in first trimester trophoblast via the TLR4/MyD88 pathway. Am J Reprod Immunol. 2009;62(2):96-111.
doi pubmed - Ninivaggi M, Kelchtermans H, Lindhout T, de Laat B. Conformation of beta2glycoprotein I and its effect on coagulation. Thromb Res. 2012;130(Suppl 1):S33-36.
doi pubmed - Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med. 2004;10(11):1222-1226.
doi pubmed - Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295-306.
doi pubmed - Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, Brey R, Crowther M, Derksen R, Erkan D, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus. 2011;20(2):206-218.
doi pubmed - Berman H, Rodriguez-Pinto I, Cervera R, Morel N, Costedoat-Chalumeau N, Erkan D, Shoenfeld Y, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab. Autoimmun Rev. 2013;12(11):1085-1090.
doi pubmed - Unlu O, Cohen H, Cuadrado M, Fortin PR, de Jesus GR, Gerosa M, Knight JK, et al. Antiphospholipid syndrome alliance for clinical trials and international networking (APS ACTION) clinical database and repository analysis: direct oral anticoagulant use among antiphospholipid syndrome patients. Presented at: 15th International Congress on Antiphospholipid Antibodies; September 21-24, 2016; Istanbul, Turkey (Relocated to North Cyprus).
- www.gov.uk/drug-safety-update/direct-acting-oral-anticoagulants-doacs-increased-risk-of-recurrent-thrombotic-events-in-patients-with-antiphospholipid-syndrome. Accessed, on April 12th, 2020.
- Pendleton R, Wheeler MM, Rodgers GM. Argatroban dosing of patients with heparin-induced thrombocytopenia and an elevated aPTT due to antiphospholipid antibody syndrome. Ann Pharmacother. 2006;40(5):972-976.
doi pubmed - Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e691S-e736S.
doi pubmed - Andrade D, Cervera R, Cohen H, Crowther M, Cuadrado MJ, Canaud G, Garcia DA, Gerosa M, et al. 15th International Congress on Antiphospholipid Antibodies Task Force on Antiphospholipid Syndrome Treatment Trends Report. In: Erkan D, Lockshin M (eds). Antiphospholipid Syndrome. Springer, Cham. 2017.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.