Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access
Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc
Journal website http://www.jocmr.org
Short Communication
Volume 9, Number 8, August 2017, pages 709-718
Splice Site Variants in the KCNQ1 and SCN5A Genes: Transcript Analysis as a Tool in Supporting Pathogenicity
Ivone U.S. Leonga, Philippa A. Drylanda, Debra O. Prossera, Stella W.-S. Laia, Mandy Grahamb, c, Martin Stilesb, c, Jackie Crawfordc, d, Jonathan R. Skinnerc, d, Donald R. Lovea, c, e
aDiagnostic Genetics, LabPlus, Auckland City Hospital, Auckland, New Zealand bDepartment of Cardiology, Waikato Hospital, Hamilton, New Zealand cCardiac Inherited Disease Group, Auckland City Hospital, Auckland, New Zealand dGreen Lane Paediatric and Congenital Cardiac Services, Starship Children’s Hospital, Auckland, New Zealand eCorresponding Author: Donald R. Love, Diagnostic Genetics, LabPlus, PO Box 110031, Auckland City Hospital, Auckland 1148, New Zealand
Manuscript submitted December 23, 2016, accepted May 22, 2017 Short title: Splice Analysis in LQT Genes doi: https://doi.org/10.14740/jocmr2894w
Background: Approximately 75% of clinically definite long QT syndrome (LQTS) cases are caused by mutations in the KCNQ1, KCNH2 and SCN5A genes. Of these mutations, a small proportion (3.2-9.2%) are predicted to affect splicing. These mutations present a particular challenge in ascribing pathogenicity.
Methods: Here we report an analysis of the transcriptional consequences of two mutations, one in the KCNQ1 gene (c.781_782delinsTC) and one in the SCN5A gene (c.2437-5C>A), which are predicted to affect splicing. We isolated RNA from lymphocytes and used a directed PCR amplification strategy of cDNA to show mis-spliced transcripts in mutation-positive patients.
Results: The loss of an exon in each mis-spliced transcript had no deduced effect on the translational reading frame. The clinical phenotype corresponded closely with genotypic status in family members carrying the KCNQ1 splice variant, but not in family members with the SCN5A splice variant. These results are put in the context of a literature review, where only 20% of all splice variants reported in the KCNQ1, KCNH2 and SCN5A gene entries in the HGMDPro 2015.4 database have been evaluated using transcriptional assays.
Conclusions: Prediction programmes play a strong role in most diagnostic laboratories in classifying variants located at splice sites; however, transcriptional analysis should be considered critical to confirm mis-splicing. Critically, this study shows that genuine mis- splicing may not always imply clinical significance, and genotype/phenotype cosegregation remains important even when mis-splicing is confirmed.
Keywords: Long QT syndrome; Splicing; Transcript analysis; In silico analysis
Long QT syndrome (LQTS) is a group of disorders characterized by a prolonged QT-interval, episodes of syncope and sudden death. The estimated number of affected people is 1 in 2,000 [1], with the peak ages of clinical presentation occurring in childhood in males, and young adult life in females, but presentation can occur at all ages, including the fetus. There are 15 genes currently associated with the disease and they mainly prolong the action potential of the cardiomyocytes by either decreasing the potassium current, through loss-of-function mutations, or increasing the sodium or calcium current through gain-of-function mutations.
LQTS subtypes 1-3 (LQTS 1-3) are the most prevalent forms of the disease and they are associated with mutations in the KCNQ1 [2], KCNH2 [3] and SCN5A [4] genes, respectively. The KCNQ1 and KCNH2 genes encode for potassium channel proteins, in which mutations account for 30-35% and 25-40% of all clinically definite LQTS cases, respectively [5]. The SCN5A gene encodes for the sodium channel Nav1.5 and mutations in this gene account for 5-10% of all LQTS cases [5]. Loss of function mutations in this gene is also associated with Brugada syndrome [6].
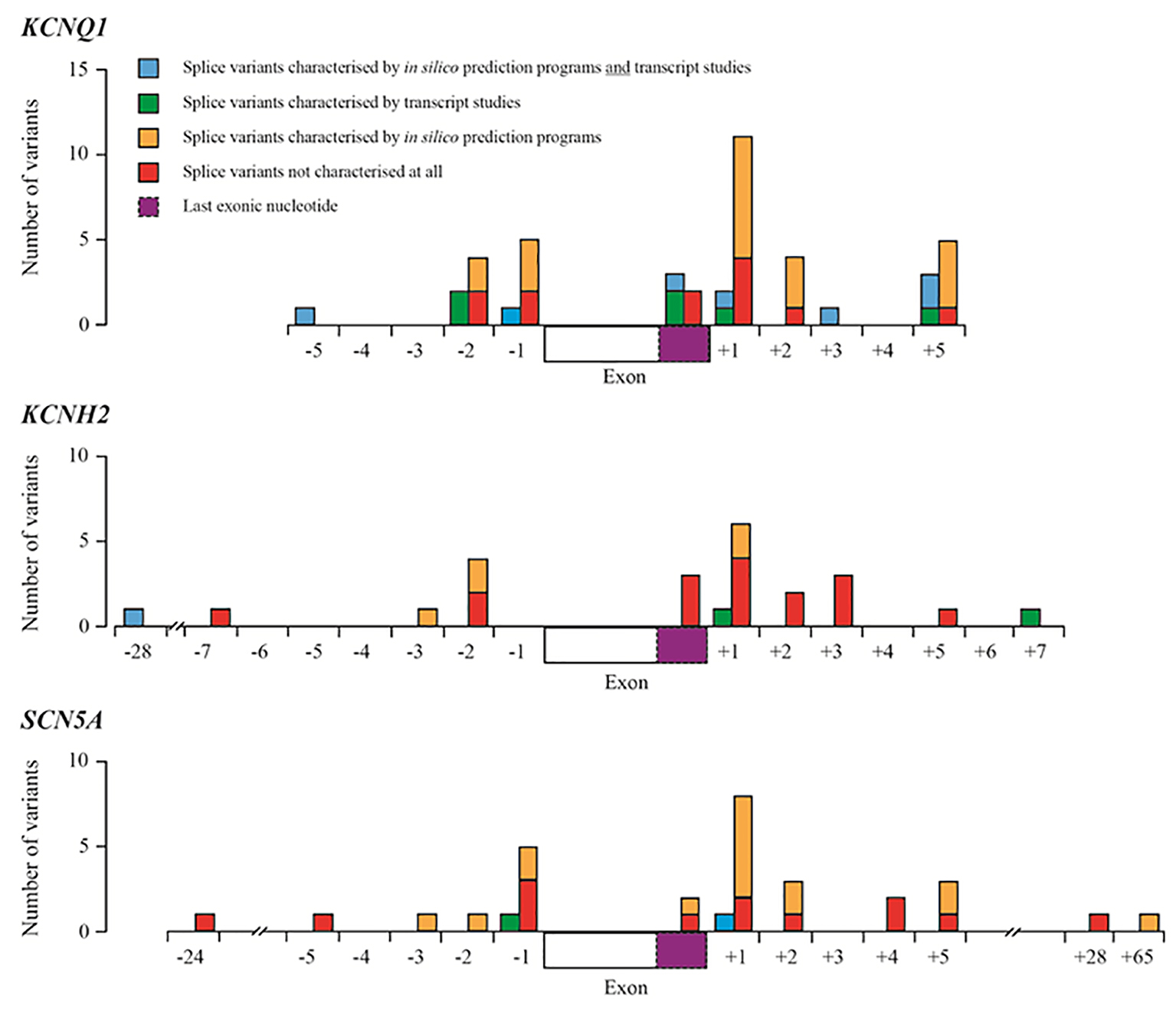
Approximately 75% of clinically definite LQTS cases are caused by mutations in the KCNQ1, KCNH2 and SCN5A genes [7, 8]. Splice site mutations in these genes make up no more than 10% of all reported mutations reported in the HGMD Pro 2015.4 database [9]. Table 1 summarizes the number and spectrum of mutations in the KCNQ1, KCNH2 and SCN5A genes. Most pathogenic mutations that affect (canonical) splicing are located at the invariant bases at 1 - 2 bp (base pairs) upstream (acceptor splice site) and downstream (donor splice site) of exon/intron boundaries; however, mutations affecting splicing have also been found to lie outside of the ± 1 - 2 bp regions [10] (Fig. 1).
Figure 1. Summary of splice variants reported in HGMD-Pro for the KCNQ1, KCNH2 and SCN5A genes. Blue boxes represent splice variants that have been characterized by in silico prediction studies and transcript studies; green boxes represent splice variants that have been characterized by transcript studies; orange boxes represent variants that have been predicted to have an effect on splicing using in silico prediction programmes (most using one programme described by Xiong et al [11]); red boxes represent splice variants that have not been characterized at all by transcript studies or in silico prediction programmes. Dashed purple boxes represent the last exonic nucleotide.
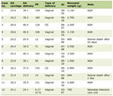
Table 1. Breakdown of Mutation Types Reported in the KCNQ1, KCNH2 and SCN5A Genes
In a diagnostic environment, methods to confirm splice-site mutations are time-consuming and presumptive splice-site mutations are normally analyzed using in silico prediction programmes. These programmes predict whether a splice-site variant is potentially pathogenic, but their efficacy varies [12]. The ideal test of whether a variant affects splicing is transcript analysis using RNA isolated from the affected tissue as cis-acting splicing mutations can have cell-specific effects [10]. In the case of cardiac genes, however, most transcript analysis involves isolating RNA from lymphocytes. Transcript analysis is not commonly performed in diagnostic laboratories so many presumptive splice-site mutations that are reported are largely not confirmed at all or are assessed only by in silico prediction programmes (Table 2).
Table 2. Breakdown of All the Splicing Mutations Reported in the KCNQ1, KCNH2 and SCN5A Genes
Here we report an analysis of the transcriptional consequences of novel genetic variants in the KCNQ1 (c.781_782delinsTC) and SCN5A (c.2437-5C>A) genes that are predicted to affect splicing in two unrelated LQTS kindreds. In both cases, transcript analysis confirmed exon skipping and a deduced translational in-frame fusion of the flanking exons. In the KCNQ1 family, the mutation co-segregates with a positive phenotype, but this is not true of the SCN5A mutation.
gDNA was extracted from peripheral blood EDTA samples using the Gentra Puregene DNA Extraction kit (Qiagen), according to the manufacturer’s instructions. This study proceeded according to informed consent and Health and Disability Ethics Committee approval (reference AKX 02/00/107).
DNA samples of the proband from families 1 and 2 were submitted to the Molecular Genetics Laboratory of The Churchill Hospital (Oxford, UK), and the Department for Medical Genetics, Oslo University Hospital (Norway), respectively, for Sanger-based sequencing analysis of the KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2 and KCNJ2 genes. Subsequent cascade Sanger-based analysis of the remaining family members was performed at Auckland City Hospital. Polymerase chain reaction (PCR) amplification of targeted exons was performed using 1× FastStart PCR buffer, 2 mM magnesium chloride, 1× GC-rich solution, 0.8 µM each of the forward and reverse primer, 0.4 mM dNTP, 0.04 U FastStart Taq DNA Polymerase (Roche), and 50 ng of genomic DNA. The following cycling conditions were used: 95 °C for 4 min, 35 cycles of 94 °C for 45 s, 60 °C for 30 s, 72 °C for 2 min 45 s, and a final extension at 72 °C for 10 min.
PCR products were treated with ExoSAP-IT (Affymetrix) prior to bi-directional DNA sequencing using BigDye Terminator v3.1 (Applied Biosystems Ltd). The sequenced products were purified using the BigDye XTerminator Purification Kit (Applied Biosystems Ltd), and then were subjected to capillary electrophoresis using the Applied Biosystems model 3130xl Genetic Analyzer. The analysis of sequence traces was performed using Variant Reporter Software 2.0 (Applied Biosystems Ltd).
Bioinformatic analysis
Berkeley Drosophila Genome Project (BDGP) Splice Site predictor [13], Alternative Splice Site Predictor (ASSP) [14], Human Splicing Finder (HSF) [15], and MaxEntScan::score5ss [16] were used to predict splicing effects. Default settings were used for all programmes.
Transcript analysis
RNA was extracted from peripheral blood EDTA samples using TRIzol reagent (Life Technologies). One microgram of total RNA was reverse transcribed to cDNA using Superscript III reverse transcriptase (Life Technologies) primed with random hexamers according to manufacturer’s instructions. cDNA quality was assessed by PCR amplification of the ABL gene transcript.
PCR amplification of defined regions of the KCNQ1 gene transcript used 1× FastStart PCR buffer, 2 mM magnesium chloride, 1× GC-rich solution, 0.8 µM each of the forward and reverse primer (Table 3), 0.4 mM dNTP, 0.04 U FastStart Taq DNA Polymerase (Roche), and 2 µL cDNA. The following cycling conditions were used: 95 °C for 4 min, 35 cycles of 94 °C for 45 s, 60 °C for 30 s, 72 °C for 2 min 45 s, and a final extension at 72 °C for 10 min.
Table 3. Sequences of Primers for the Analysis of Gene Transcripts in Families 1 and 2
In the case of the SCN5A gene transcript, a nested PCR approach was used. The initial PCR comprised 1× FastStart PCR buffer, 2 mM magnesium chloride, 1× GC-rich solution, 0.8 µM each of the forward and reverse outer primer (Table 3), 0.4 mM dNTP, 0.04 U FastStart Taq DNA Polymerase (Roche), and 2 µL cDNA. The following cycling conditions were used: 95 °C for 4 min, 30 cycles of 94 °C for 45 s, 60 °C for 30 s, 72 °C for 2 min 45 s, and a final extension at 72 °C for 10 min. The secondary (nested) PCR mixture comprised 1 µL of the first PCR product, together with 1× FastStart PCR buffer, 2 mM magnesium chloride, 1× GC-rich solution, 0.8 µM each of the forward and reverse inner primer (Table 3), 0.4 mM dNTP, and 0.04 U FastStart Taq DNA Polymerase (Roche). The following cycling conditions were used: 95 °C for 4 min, 30 cycles of 94 °C for 45 s, 60 °C for 30 s, 72 °C for 2 min 45 s, and a final extension at 72 °C for 10 min.
All PCR amplicons were visualized by electrophoresis in 2% EX Agarose Gels (Life Technologies) and amplicons were extracted from gels using the QIAquick Gel Extraction Kit (Qiagen) according to the manufacturer’s instructions. Bi-directional DNA sequencing was performed using BigDye Terminator v3.1 (Applied Biosystems Ltd). The sequenced products were purified using the BigDye XTerminator Purification Kit (Applied Biosystems Ltd), and then were subjected to capillary electrophoresis using an Applied Biosystems model 3130xl Genetic Analyzer. The analysis of sequence traces was performed using Geneious v7.1.7 software (Biomatters) [17].
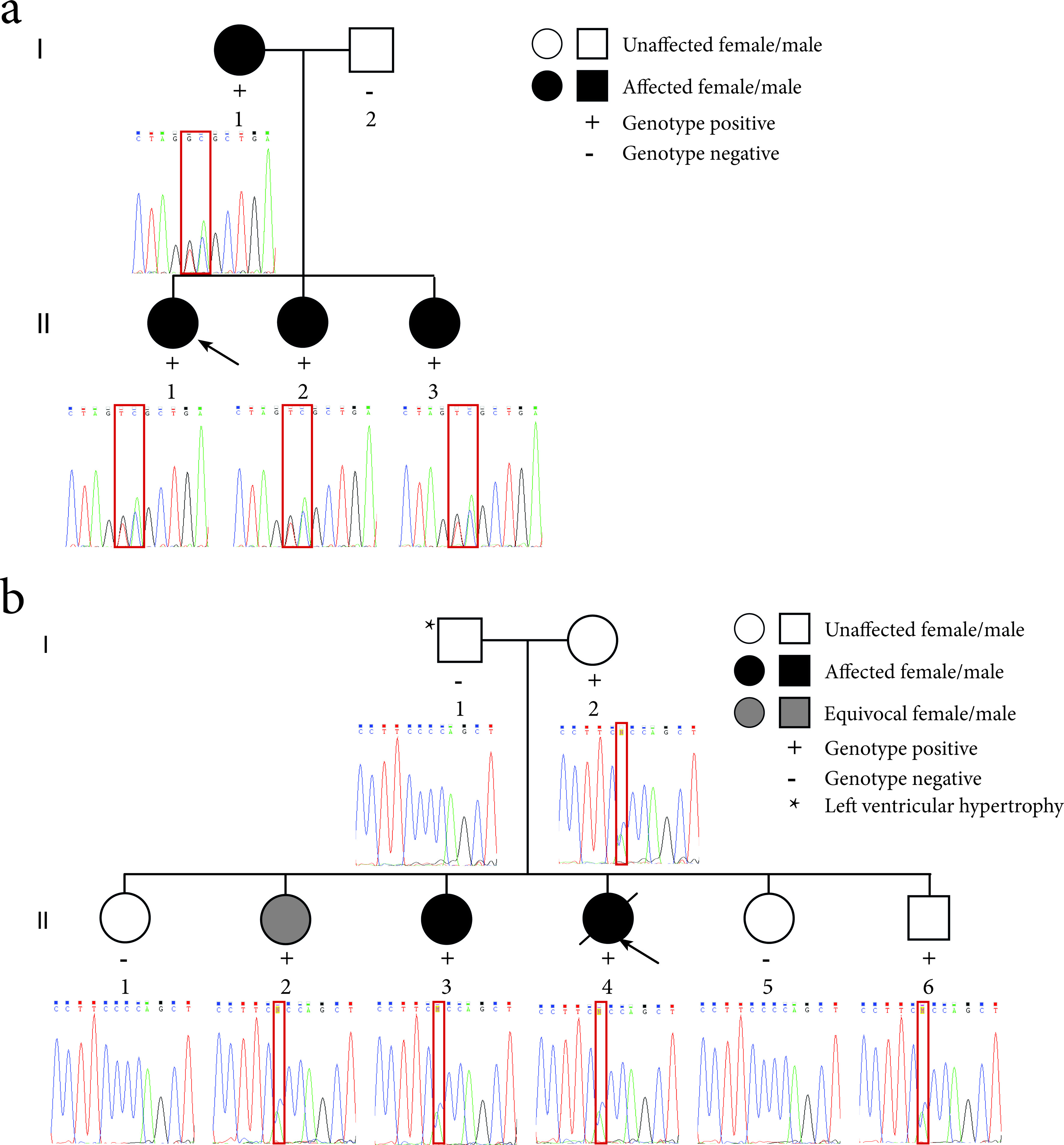
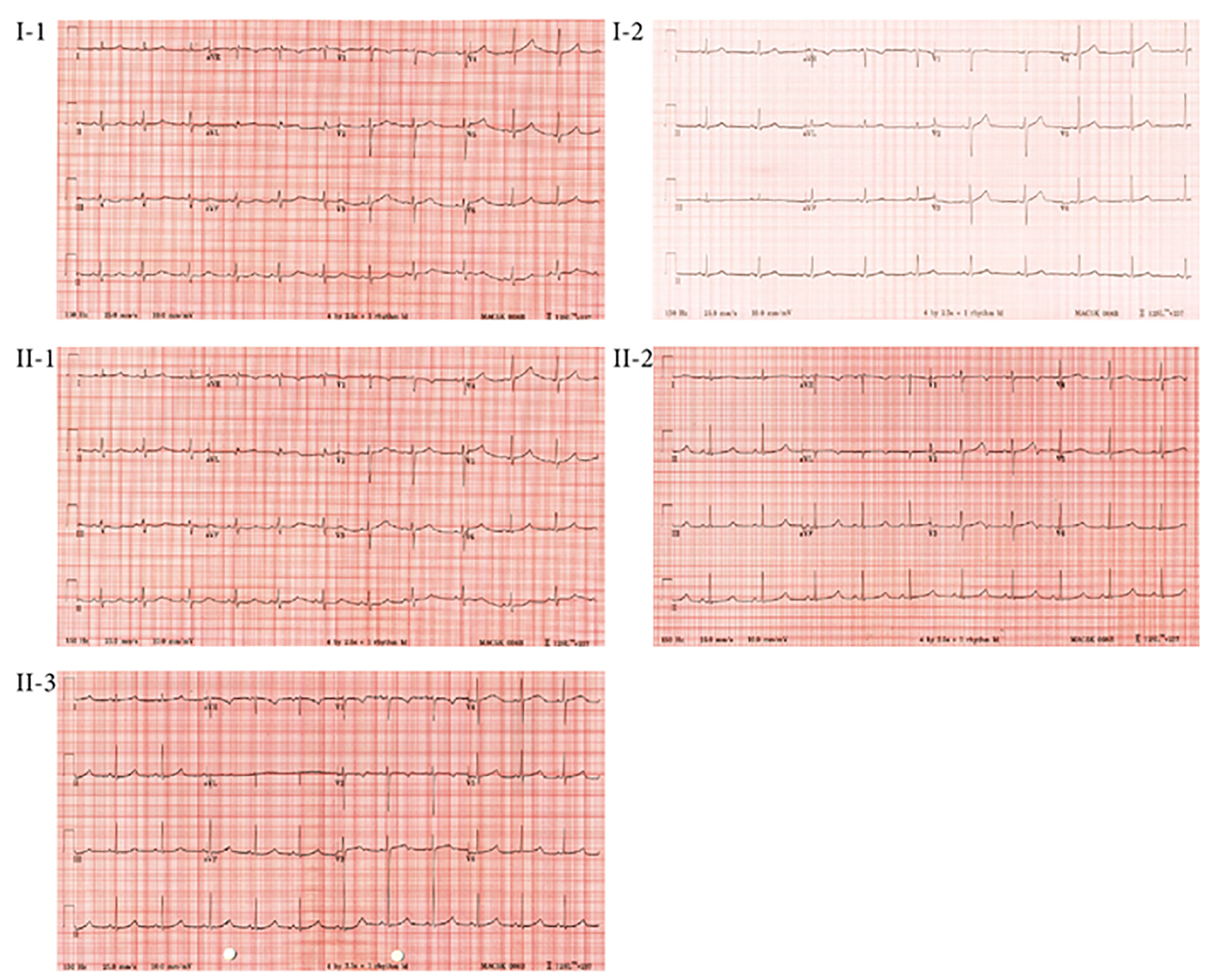
Family 1 (Fig. 2a): the proband (II-1) was a 15-year-old girl with no prior medical history who presented with syncope after running up a flight of stairs. She was referred to the national Cardiac Inherited Disease Group (CIDG) for further cardiac/genetic investigation. The proband’s maternal grandfather suffered sudden death at 39 years of age, and two of the grandfather’s three siblings also died suddenly at a young age (10 years, during a running race, and 21 years, post routine surgery). The proband’s cardiovascular examination was normal. A 12-lead ECG of the proband showed a QTc of 530 ms, with broad-based, late-onset T-waves (Fig. 3). She was clinically diagnosed with LQTS, commenced on nadolol and had a left cardiac sympathetic denervation (LCSD).
Figure 2. Pedigrees of the LQTS families and sequence electropherograms. (a) Family 1 segregates the c.781_782delinsTC mutation in the KCNQ1 gene. The proband (II-1) is indicated by the black arrow. (b) Family 2 segregates the c.2437-5C>A mutation in the SCN5A gene. The proband (II-4) is indicated by the black arrow. The location of the mutation in the sequence electropherograms is indicated by a red box.
The proband’s mother’s (I-1) ECG showed a QTc of 475 ms and the T-wave morphology was suggestive of LQTS 1 (Fig. 3). The proband’s father (I-2) had a normal ECG (QTc of 424 ms; Fig. 3). The proband’s two younger asymptomatic sisters (II-2, aged 11 years; II-3, aged 7 years) had QTcs of 588 and 521 ms, respectively, and both had late onset T-waves (Fig. 3). They were commenced on medical therapy, and II-2 also proceeded to LCSD. All family members have thus far remained asymptomatic.
Family 2 (Fig. 2b): the proband (II-4) was a 25-year-old woman who died in her sleep 4 months post-partum, after a 6-day illness (including seizures) thought to be due to food poisoning. Thorough autopsy investigation, including detailed cardiac investigation, revealed no cause for death. Her family was referred to the CIDG for further cardiac/genetic investigation according to local protocols [18]. No significant family history was apparent and no ECG of the proband existed. Her parents had normal echocardiography and cardiac MR scans and her father exhibited mild left ventricular hypertrophy. Investigation by ECG and/or Holter monitoring revealed normal QTc intervals for I-2 and II-6, both with normal nocturnal QT behavior, but II-2 had a borderline QTc of 465ms and a history of a single startle related syncope, and II-3 had a distinctly prolonged QTcs of 490 ms, both with borderline QTc prolongation at night (nocturnal QTc prolongation being a feature of LQT3, which is linked to SCN5A).
Molecular genetic analysis
Family 1
Sanger-based sequence analysis of the proband (II-1) identified two apparently single heterozygous mutations in the KCNQ1 gene: c.781G>T and c.782A>C (Fig. 2a). The proband’s sisters and her mother also carried the mutations, suggesting that the mutations are in cis and so should be reported as c.781_782delinsTC. The predicted effect of this mutation at the amino acid level suggested an amino acid change of glutamic acid to serine at residue 261. Alternatively, at the transcript level, in silico splice site analysis programmes predicted that the mutation, located at the proximal (5′) end of exon 6, would abolish the splice acceptor site of exon 6 and hence lead to exon skipping.
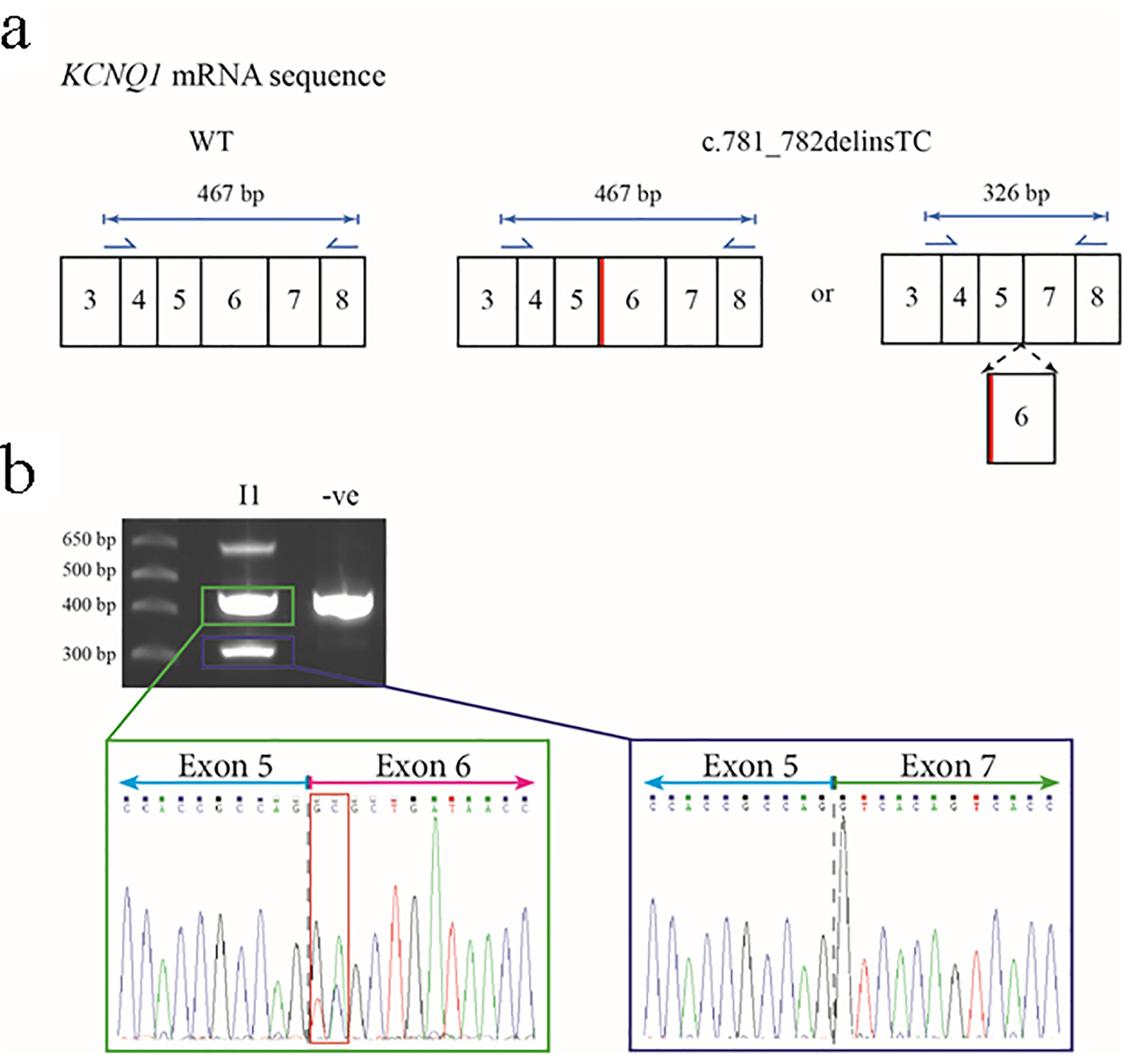
Transcript analysis was performed on the proband’s mother (I-1) to resolve the above predictions. Primers were designed to amplify regions of the KCNQ1 gene transcript encompassing exon 6 (Fig. 4a). Sanger-based sequencing of the about 470 bp and about 330 bp products revealed that the latter corresponded to the predicted mis-spliced (loss of exon 6) product while the former corresponded to canonical splicing with no loss of exon 6 (Fig. 4b).
Figure 4. Predicted outcomes for carriers of the c.781_782delinsTC mutation in the KCNQ1 gene, together with transcript analysis. (a) The wild-type (WT) KCNQ1 gene transcript would be unaffected and produce normal KCNQ1, but the mutation could cause an amino acid change at residue 261 of glutamic acid to alanine (p.E261A) or cause exon 6 to be spliced out. Primers for the amplification of cDNA are shown as blue arrows with the lengths of anticipated amplicons shown above the relevant primer pairs. The diagram only shows a partial representation of the KCNQ1 mRNA sequence instead of all 16 exons. The thick red line shows the location of the c.781_782delinsTC mutation in relation to the rest of the exons. (b) The 2% agarose gel showing the results of the PCR amplification of cDNA from the proband’s mother (I-1) and an unrelated control. The highest molecular weight product is a heteroduplex of the two smaller amplicons (representing non-excised and excised exon 6 transcripts). The sequence electropherogram of the about 470 bp amplicon (green box) shows the sequence of the junction between exons 5 and 6 (gray dashed line) and heterozygosity for the c.781_782delinsTC mutation (red box). The approximate 330 bp amplicon (blue box) shows the in-frame fusion of exon 5 and exon 7 (gray dashed line).
Family 2
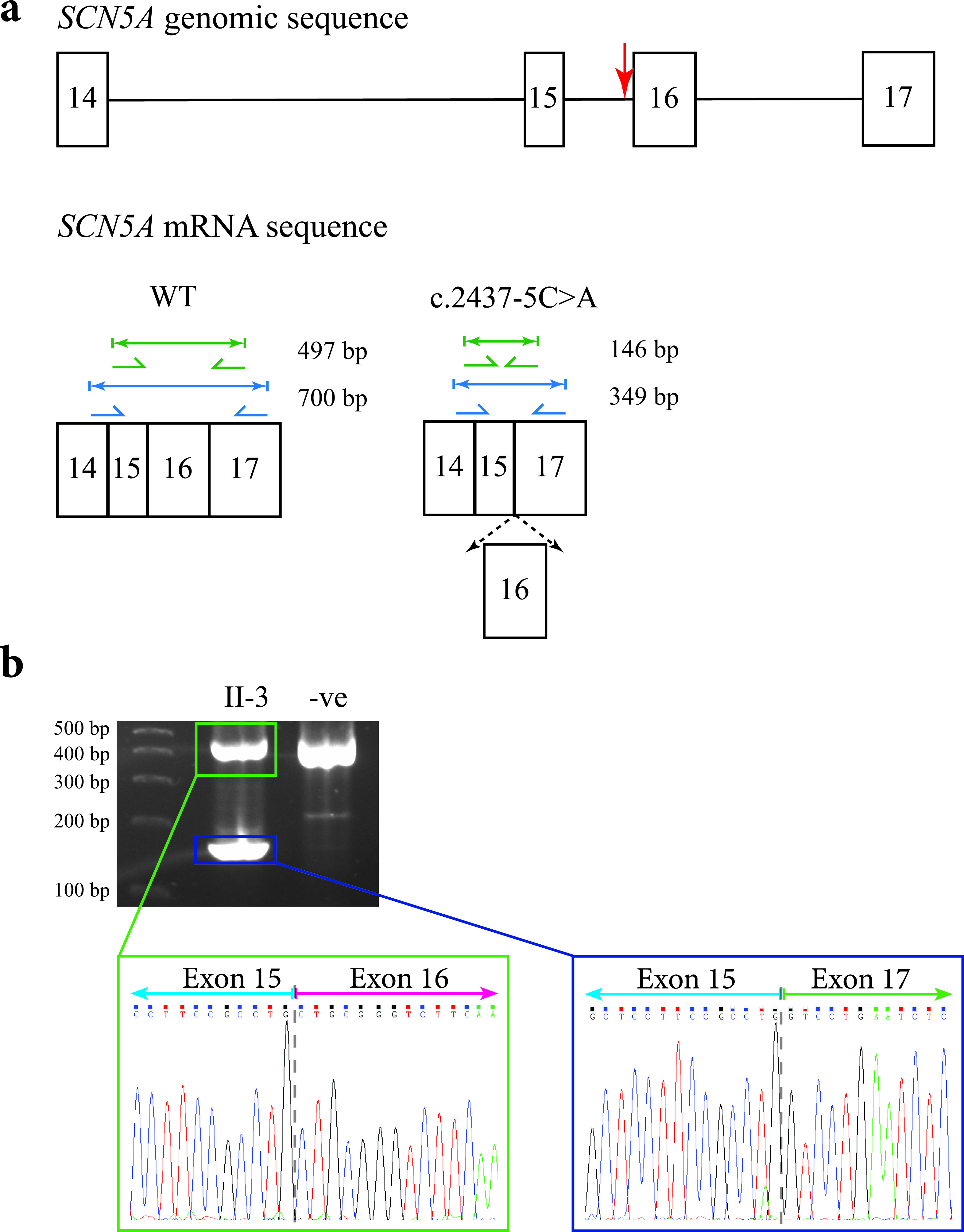
Sanger-based sequence analysis of the deceased proband (II-4), as well as the proband’s mother (I-2), two sisters (II-2 and II-3) and brother (II-6) indicated that they carried a heterozygous mutation in the SCN5A gene: c.2437-5C>A (Fig. 2b). This mutation lies upstream of the splice acceptor site of exon 16. In silico splice site analysis predicted that the mutation would abolish the function of the splice acceptor site leading to the loss of exon 16, and a subsequent in-frame fusion of exons 15 and 17 (Fig. 5a).
Figure 5. Predicted outcomes for carriers of the c.2437-5C>A mutation in the SCN5A gene, together with transcript analysis. (a) Diagrammatic representation of the genomic location of the c.2437-5C>A mutation in the SCN5A gene in relation to the other exons (indicated by the red arrow; top), and the amplicon sizes of the expected PCR products for an unaffected individual (bottom left), and the shortened amplicon sizes for the carriers of the mutation if exon 16 were spliced out (bottom right). The diagram only shows a partial representation of the SCN5A genomic/mRNA sequence instead of all 16 exons. (b) The 2% agarose gel showing the results of PCR amplification of cDNA from the proband’s sister (II-3) and an unrelated control. The sequence electropherogram of the higher molecular weight amplicon (green box) shows the junction between exons 15 and 16 (gray dashed line). The sequence electropherogram of the lower molecular weight amplicon (blue box) shows the in-frame fusion of exons 15 and 17 (gray dashed line).
Nested PCR amplification was performed using lymphocyte RNA isolated from the proband’s mother (I-2) and two sisters (II-2 and II-3). Sanger-based sequencing of the smaller amplicon confirmed the predicted mis-splicing and loss of exon 16 (Fig. 5b).
Given the recognized cross over phenotype seen with SCN5A gene mutations, all three gene-positive siblings of the deceased proband underwent sodium channel blockade with Ajmaline and none revealed a Brugada ECG signature.
The c.781_782delinsTC mutation in the KCNQ1 gene has not been reported previously in any databases. Interestingly, single mutations have been reported at each of the 781 and 782 nucleotide positions. A c.781G>T mutation has been reported by Kapplinger et al [7] where it was found in one of 2,500 unrelated patients with a subsequent prediction that it would have a splicing effect [8]. A c.781G>C mutation (p.Glu261Gln; rs199472722) has been found in 2,500 unrelated patients and is predicted to cause a missense amino acid change as well as affect splicing [7]. Finally, c.781G>A (p.Glu261Lys; rs199472722) has been investigated by three groups [19, 20], and appears to cause a loss-of-function of the mutant protein [20, 21].
A c.782A>C mutation (p.Glu261Val) has not been previously reported in any databases; however, a nucleotide change of A>T has been reported at this site and the same patient also has the c.781G>T mutation [22]. No further details were given for this patient so it is not clear whether the two mutations are in cis or in trans, and no transcript analysis was performed.
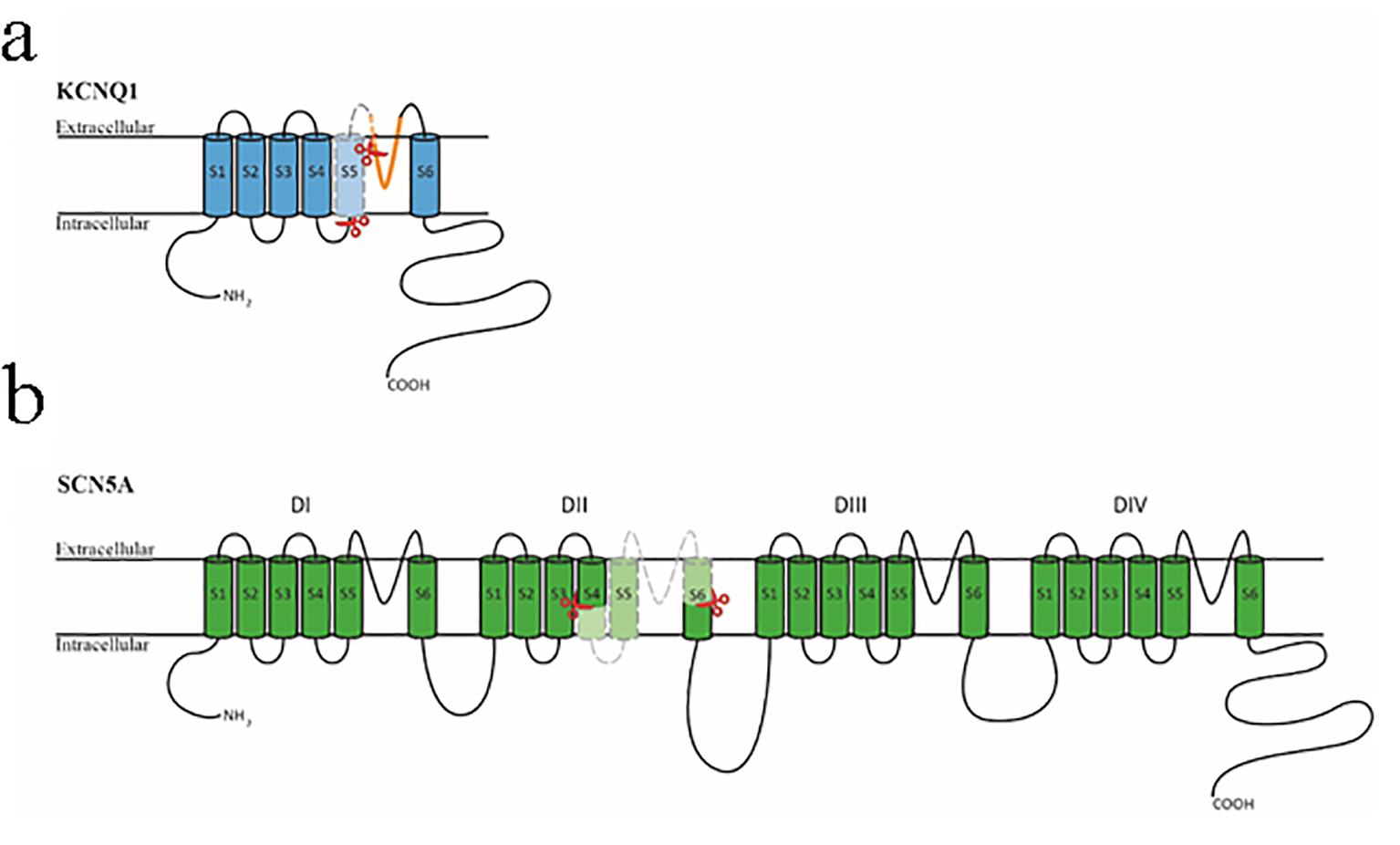
The c.781_782delinsTC mutation is predicted to affect the correct splicing of the KCNQ1 gene transcript, which has been confirmed here. The KCNQ1 protein is composed of six transmembrane domains (S1-S6) with the pore region between the S5 and S6 domains (Fig. 6a). Exon 6 of the KCNQ1 gene transcript encodes S5 and part of the pore domain of the KCNQ1 protein [23], and 58 missense, small deletions and splice mutations associated with LQTS 1 are located in this region [9]. Many of the missense mutations have been functionally characterized and have been shown to affect KCNQ1 protein function, therefore the splicing out of exon 6 of the KCNQ1 gene transcript is expected to be deleterious. The proband’s mother and two sisters carry the same mutation and exhibit signs of a prolonged QT-interval and late onset T-wave. Taken together, our data support the c.781_782delinsTC mutation in the KCNQ1 gene to be disease-causing.
Figure 6. Diagrammatic representation of the KCNQ1 and SCN5A proteins. (a) The KCNQ1 protein is composed of six transmembrane domains (S1-S6) with a pore region between S5 and S6 (shown in orange). The section encompassed by the two red scissors and colored in light blue is encoded by exon 6 of the KCNQ1 gene. (b) The SCN5A protein is composed of four homologous transmembrane domains (DI-DIV) with six transmembrane segments (S1-S6) in each section. The four sections’ S1-S4 domains form the channel’s voltage-sensing region, and the four S5-S6 domains with the intervening loop region form the central pore region and selective filter. The section encompassed by the two red scissors and colored in light green is encoded by exon 16 of the SCN5A gene.
The c.2437-5C>A mutation (rs72549411) in the SCN5A gene has been reported as benign in the ClinVar database [24]; however, there are no publications regarding this mutation. There are also no known mutations in this region that affect splicing. Splice prediction programmes suggest that this mutation would cause mis-splicing of the SCN5A gene transcript, which has been confirmed here. The SCN5A protein is composed of four repetitive transmembrane domains (DI-DIV) with six transmembrane segments (S1-S6) for each section (Fig. 6b). The S1-S4 domains form the sodium channel’s voltage-sensing domain, in which the positively charged S4 domain is vital for its function [25]. The S5-S6 domains and the intervening loop region form the central pore region and selectivity filter [26]. Exon 16 of SCN5A gene encodes for transmembrane domains 4-6 and the loop region of DII (Fig. 6b). There are 39 missense and small deletion mutations associated with LQTS 3 located in this region [9], and some of the missense mutations have been found to be deleterious through functional studies. Aside from the deceased proband one gene-positive sibling showed an equivocal long QTc interval and a possible arrhythmic syncope but two mutation-positive family members were asymptomatic with normal QTc intervals and ajmaline challenges. Overall, this variant should be classified as of unknown clinical significance despite the detectable exon-skipping. Functional studies of the mutant SCN5A protein may resolve this classification.
The variable efficacy of splice site prediction programmes underscores the need for transcript studies to provide biological reality to in silico conjecture. In both cases reported here, the predictions were verified, but protein domain and segregation analysis coupled with ECG data were required to resolve the classification of variants as clinically actionable, or not.
Of the 95 splice variants in the KCNQ1, KCNH2 and SCN5A genes reported in the HGMDPro 2015.4 database, approximately 20% have been verified by transcript analysis as leading to mis-splicing, with the majority (70%) reported in the KCNQ1 gene (Tables 1 and 2). Interestingly, in silico programmes were used to predict pathogenicity for twice as many splice variants as those that were biologically verified by transcript studies. Of the 18 transcript-verified variants, approximately half are located at the invariant bases of the acceptor and donor splice sites (± 1 - 2 bp from exon/intron boundaries) while the remainder are either located within an exon or lie further away from the invariant bases. These data suggest that transcript analysis is not a common tool to aid in the classification of apparent splice variants, and that there is a strong preference to use prediction programmes that are less reliable in classifying variants that lie outside the invariant bases [10].
There are only two other reported transcript studies supporting pathogenicity for apparent splice site variants in the SCN5A gene, and our single case reported here is the third, and only one lying outside the invariant bases. The lack of genotypic correlation with phenotype in our SCN5A kindred may signal a need for extreme caution in interpreting such variants in a gene that is linked to different cardiac phenotypes.
Conclusions
We report two unrelated cases of mutations in the KCNQ1 and SCN5A genes that are predicted to affect splicing. Transcript analysis performed on both families confirmed the predicted mis-splicing and cause an exon of each gene to be excised resulting in an in-frame fusion of the flanking exons. Despite this finding, further work may be required at the protein level to determine the role that in-frame fusions have on protein function. Generally, however, our results highlight the importance of undertaking two inter-related analyses for splice-site variants. The first, transcript analysis, should be used to provide biological evidence of mis-splicing. The second, segregation analysis, should allow genotype: phenotype correlations to be made.
Acknowledgments
We thank the families for kindly permitting this report and Charlene Nell, Desktop Support Administrator, for preparing the manuscript and for excellent secretarial assistance.
Financial Disclosure
The study was financially supported by the National Heart Foundation (New Zealand), the Auckland Medical Research Foundation, the Lottery Grants Board of New Zealand and the Maurice and Phyllis Paykel Trust. The Cardiac Inherited Disease Group is supported by Cure Kids, who also partially fund Dr Skinner’s and Dr Leong’s salaries. Dr Leong was financially supported during a large part of this work by The Rutherford Foundation as a New Zealand Postdoctoral Fellow.
Conflicts of Interest
The authors have no conflicts of interest to declare, and alone are responsible for the content and writing of the paper.
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761-1767. doipubmed
Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17-23. doipubmed
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795-803. doi
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80(5):805-811. doi
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308-1339. doipubmed
Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, et al. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2(4):429-440. doipubmed
Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6(9):1297-1303. doipubmed
Earle N, Crawford J, Smith W, Hayes I, Shelling A, Hood M, Stiles M, et al. Community detection of long QT syndrome with a clinical registry: an alternative to ECG screening programs? Heart Rhythm. 2013;10(2):233-238. doipubmed
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1-9. doipubmed
Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. 2005;42(10):737-748. doipubmed
Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. doipubmed
Tang R, Prosser DO, Love DR. Evaluation of bioinformatic programmes for the analysis of variants within splice site consensus regions. Advances in Bioinformatics. 2016;2016:1-10. doipubmed
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311-323. doipubmed
Wang M, Marin A. Characterization and prediction of alternative splice sites. Gene. 2006;366(2):219-227. doipubmed
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. doipubmed
Eng L, Coutinho G, Nahas S, Yeo G, Tanouye R, Babaei M, Dork T, et al. Nonclassical splicing mutations in the coding and noncoding regions of the ATM Gene: maximum entropy estimates of splice junction strengths. Hum Mutat. 2004;23(1):67-76. doipubmed
Biomatters. GeneGene vR6.1.6; Available from: http://www.geneious.com/
Skinner JR, Crawford J, Smith W, Aitken A, Heaven D, Evans CA, Hayes I, et al. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm. 2011;8(3):412-419. doipubmed
Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M, Chivoret G, et al. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation. 1997;96(9):2778-2781. doipubmed
Wilson AJ, Quinn KV, Graves FM, Bitner-Glindzicz M, Tinker A. Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1). Cardiovasc Res. 2005;67(3):476-486. doipubmed
Franqueza L, Lin M, Shen J, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J Biol Chem. 1999;274(30):21063-21070. doipubmed
Yoshinaga M, Kucho Y, Sarantuya J, Ninomiya Y, Horigome H, Ushinohama H, Shimizu W, et al. Genetic characteristics of children and adolescents with long-QT syndrome diagnosed by school-based electrocardiographic screening programs. Circ Arrhythm Electrophysiol. 2014;7(1):107-112. doipubmed
Cui J. Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophys J. 2016;110(1):14-25. doipubmed
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862-868. doipubmed
Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108(7):884-897. doipubmed
Terlau H, Heinemann SH, Stuhmer W, Pusch M, Conti F, Imoto K, Numa S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293(1-2):93-96. doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.