

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 9, Number 4, April 2017, pages 233-247
Insulin-Like Growth Factor-1 Deficiency and Cirrhosis Establishment
Rocio G. de la Garzaa, Luis Alonso Morales-Garzaa, Irene Martin-Estala, Inma Castilla-Cortazara, b, c
aCentro de Investigacion Transferencia en Salud (CITES), Escuela Nacional de Medicina, Tecnologico de Monterrey, and Institute of Liver Diseases, Hospital San Jose, Tecnologico de Monterrey, Monterrey, Nuevo Leon, Mexico
bFundacion de Investigacion HM Hospitales, Madrid, Spain
cCorresponding Author: Inma Castilla-Cortazar, Escuela Nacional de Medicina, Tecnologico de Monterrey, Avenida Morones Prieto 3000 Pte. Col. Los Doctores, 64710, Monterrey, Nuevo Leon, Mexico
Manuscript accepted for publication October 19, 2016
Short title: IGF-1 Deficiency and Liver Disease
doi: https://doi.org/10.14740/jocmr2761w
- Abstract
- Introduction
- Epidemiology
- Growth Hormone (GH)-IGF-1 Axis, IGF-1 Deficiency, Altered Lipid Metabolism and Oxidative Damage
- Physiopathological Pathways Since Early Liver Damage to Decompensated Cirrhosis: Relationship With IGF-1 Deficiency
- IGF-1 and GH in Cirrhosis
- Results of IGF-1 Therapy
- Other Strategies for Cirrhosis Treatment
- Conclusions and Future Perspective
- References
| Abstract | ▴Top |
Cirrhosis represents the final stage of chronic liver damage, which can be due to different factors such as alcohol, metabolic syndrome with liver steatosis, autoimmune diseases, drugs, toxins, and viral infection, among others. Nowadays, cirrhosis is an important health problem and it is an increasing cause of morbidity and mortality, being the 14th most common cause of death worldwide. The physiopathological pathways that lead to fibrosis and finally cirrhosis partly depend on the etiology. Nevertheless, some common features are shared in this complex mechanism. Recently, it has been demonstrated that cirrhosis is a dynamic process that can be altered in order to delay or revert fibrosis. In addition, when cirrhosis has been established, insulin-like growth factor-1 (IGF-1) deficiency or reduced availability is a common condition, independently of the etiology of chronic liver damage that leads to cirrhosis. IGF-1 deprivation seriously contributes to the progressive malnutrition of cirrhotic patient, increasing the vulnerability of the liver to establish an inflammatory and oxidative microenvironment with mitochondrial dysfunction. In this context, IGF-1 deficiency in cirrhotic patients can justify some of the common characteristics of these individuals. Several studies in animals and humans have been done in order to test the replacement of IGF-1 as a possible therapeutic option, with promising results.
Keywords: IGF-1; Steatosis; Non-alcoholic fatty liver disease; Acute liver damage; GH/IGF-1 axis; Fibrogenesis; Oxidative damage; Mitochondrial protection
| Introduction | ▴Top |
Cirrhosis is the result of chronic liver disease. It represents the final stage of a wide number of chronic liver conditions, whose common effect is necroinflammation, fibrosis and regeneration nodules that modify the normal liver structure reducing its functional mass and altering the vascular liver architecture [1-3]. Fibrosis progresses at variable rates depending on the cause of liver disease, environmental and host factors [4-6]. However, all those structural liver changes lead to impaired hepatocyte function (hepatocellular insufficiency) and an increased intrahepatic pressure (portal hypertension) leading to all the clinical manifestations in cirrhosis [7].
The transition from chronic liver disease to cirrhosis involves inflammation, and activation of hepatic stellate cells (HSCs) leading to fibrogenesis - angiogenesis and parenchymal lesions - partly due to vascular occlusion (Fig. 1) [8]. These changes produce hepatic microvascular rearrangement, such as sinusoidal remodeling, formation of intrahepatic shunts, and hepatic endothelial dysfunction [9]. The endothelial dysfunction alters the normal release of vasodilators, most importantly nitric oxide. In addition, there is an increased production of vasoconstrictors [10]. All these changes, in combination with structural disturbances generate increased hepatic resistance to portal blood flow, leading to an elevated portal pressure, with its clinical consequences.
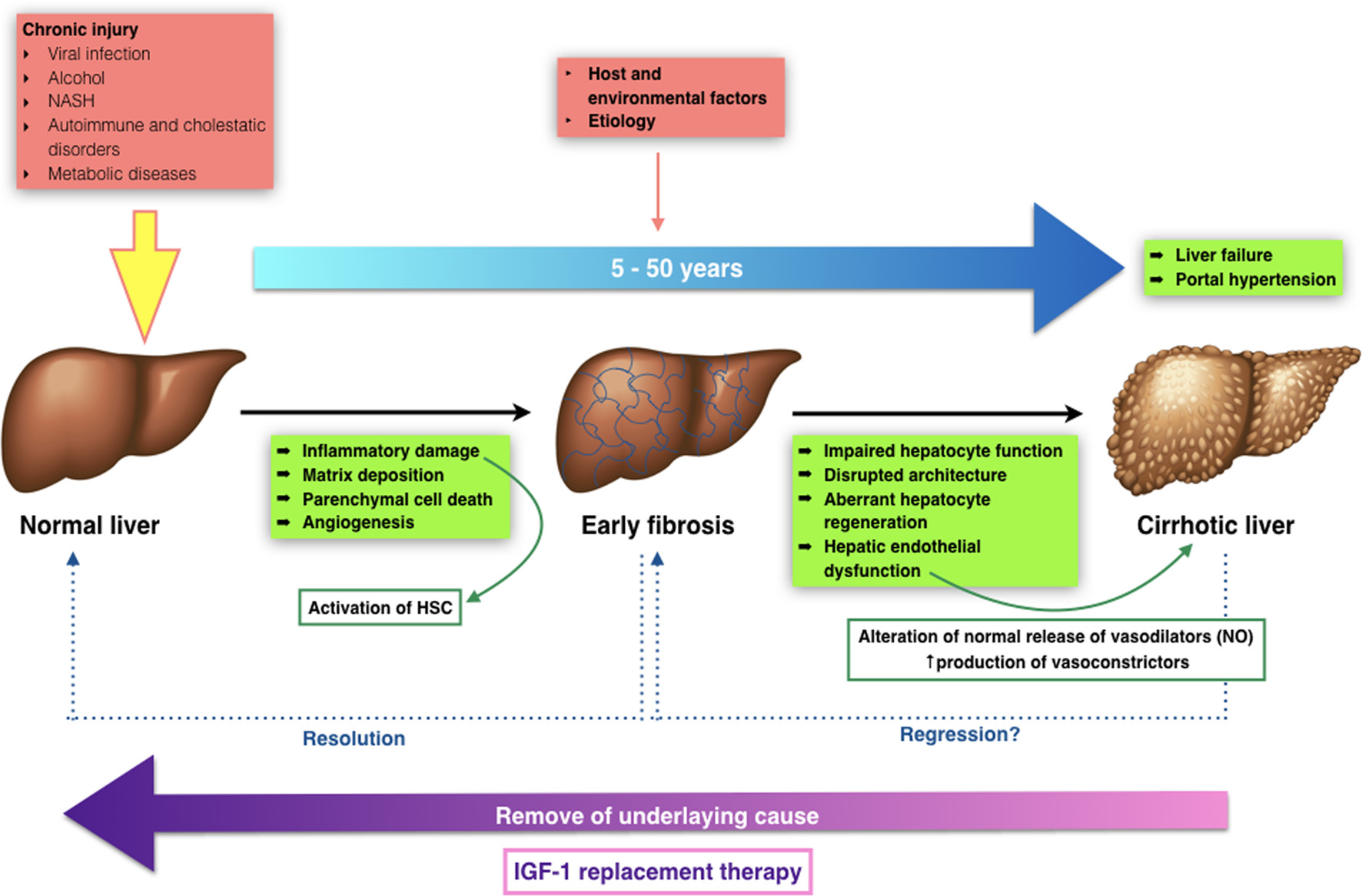 Click for large image | Figure 1. Transition from normal to liver cirrhosis. |
Furthermore, recently our group has described that the mere insulin-like growth factor-1 (IGF-1) partial deficiency, in an animal model with such deficiency, is associated with relevant alterations of hepatic architecture and expression of genes involved in cytoskeleton, hepatocyte polarity, cell junction and extracellular matrix (ECM) proteins [11]. These results suggest a novel approach to overcome the physiopathology in the onset of liver damage, IGF-1 availability, IGF-1 receptor expression after injury and cirrhosis development, claiming further investigation. To date, such results have not been studied in humans.
Cirrhosis is a dynamic process that has to be monitored frequently, in order to avoid progression and/or reverse fibrosis [12, 13]. It can remain in a compensated state for several years, but when progression persues to advanced stages, complications may appear leading to a poor quality of life with higher morbidity and mortality. In advanced stages, the most common complications include coagulopathy and jaundice, gastrointestinal bleeding from esophageal varices, ascites, hepatorenal syndrome, spontaneous bacterial peritonitis, encephalopathy, hipogonadism, and malnutrition [14, 15].
Nowadays, when treating cirrhotic patients, the aim is to avoid or delay progression to a “decompensated” stage, where mortality rises up to 85% over 5 years, and to avoid liver transplantation [7]. Nevertheless, drug therapies can partially prevent or control some complications, but none of them can significantly increase survival nor modify the natural clinical course of the disease, except for some etiologic specific therapies.
For this reason, new therapies have been tested in order to modify the natural history of cirrhosis, improving hepatocellular function and reducing portal pressure. In this context, it seems necessary to improve our knowledge in the early onset of liver disease as well as its transition to cirrhosis, in order to find different therapeutic alternatives. This approach will be reviewed in the following lines.
| Epidemiology | ▴Top |
Cirrhosis is an increasing cause of morbidity and mortality, being the 14th most common cause of death worldwide [7]. Its prevalence is difficult to assess because the initial stages of the disease are asymptomatic, thus remaining undiagnosed, and is probably higher than reported.
In a recent epidemiologic study of cirrhosis, global liver cirrhosis deaths represented more than one million in 2010, or 1.95% of the global total deaths. On average, there were twice as much male deaths compared to women [16].
Furthermore, most Western European countries have improved its cirrhosis mortality, with the exception of UK, Ireland and Finland, where cirrhosis mortality rates have been increasing since 1980 [16]. In Latin America, mortality rates vary among different countries. Mexico has remained the country with the highest cirrhosis mortality rate in the region (Latin America), at 38.3 (30.7 - 47.5) per 100,000, and in 2010, it was the fourth leading cause of death, accounting for 18% of deaths in males aged 40 - 49 years [16]. Concerning Central Asia, the mortality rates have increased since 1990s until 2000, but in the last decade, the mortality rate has decreased or stabilized in these countries. Finally, in sub-Saharan Africa, cirrhosis deaths have been doubled between 1980 and 2010 [16].
Alcoholic liver disease and hepatitis C are the main causes in most developed countries, while hepatitis B is the most common cause in most parts of Asia and sub-Saharan Africa (Table 1) [3, 16].
 Click to view | Table 1. Population Fractions for Liver Cirrhosis Risk Factors by Region in 2010 |
Moreover, it is well known that metabolic syndrome (MetS) is increasing worldwide [17], in part related to the occidentalization of lifestyle habits [18, 19]. Non-alcoholic liver disease is also increasing in developed countries [20, 21]. It is important to take this into account, as MetS represents a major cause of non-alcoholic steatohepatitis (NASH) and non-alcoholic fatty liver disease (NAFLD). Of interest, accumulated evidence relates IGF-1 deficiency with MetS establishment and steatohepatitis, since the mere IGF-1 deficiency alters hepatic expression of gene involved in glucose and lipid metabolism [22].
| Growth Hormone (GH)-IGF-1 Axis, IGF-1 Deficiency, Altered Lipid Metabolism and Oxidative Damage | ▴Top |
IGF-1 is a 70-aminoacidic anabolic hormone with several endocrine, paracrine and autocrine effects [23]. It is well known that IGF-1 is mainly produced by the liver (accounting for 75% of circulating IGF-1), but almost every tissue is able to secrete IGF-1 for autocrine/paracrine purposes [24-27].
Pituitary GH and liver derived IGF-1 establish a negative feedback mechanism (Fig. 2) [28-30].
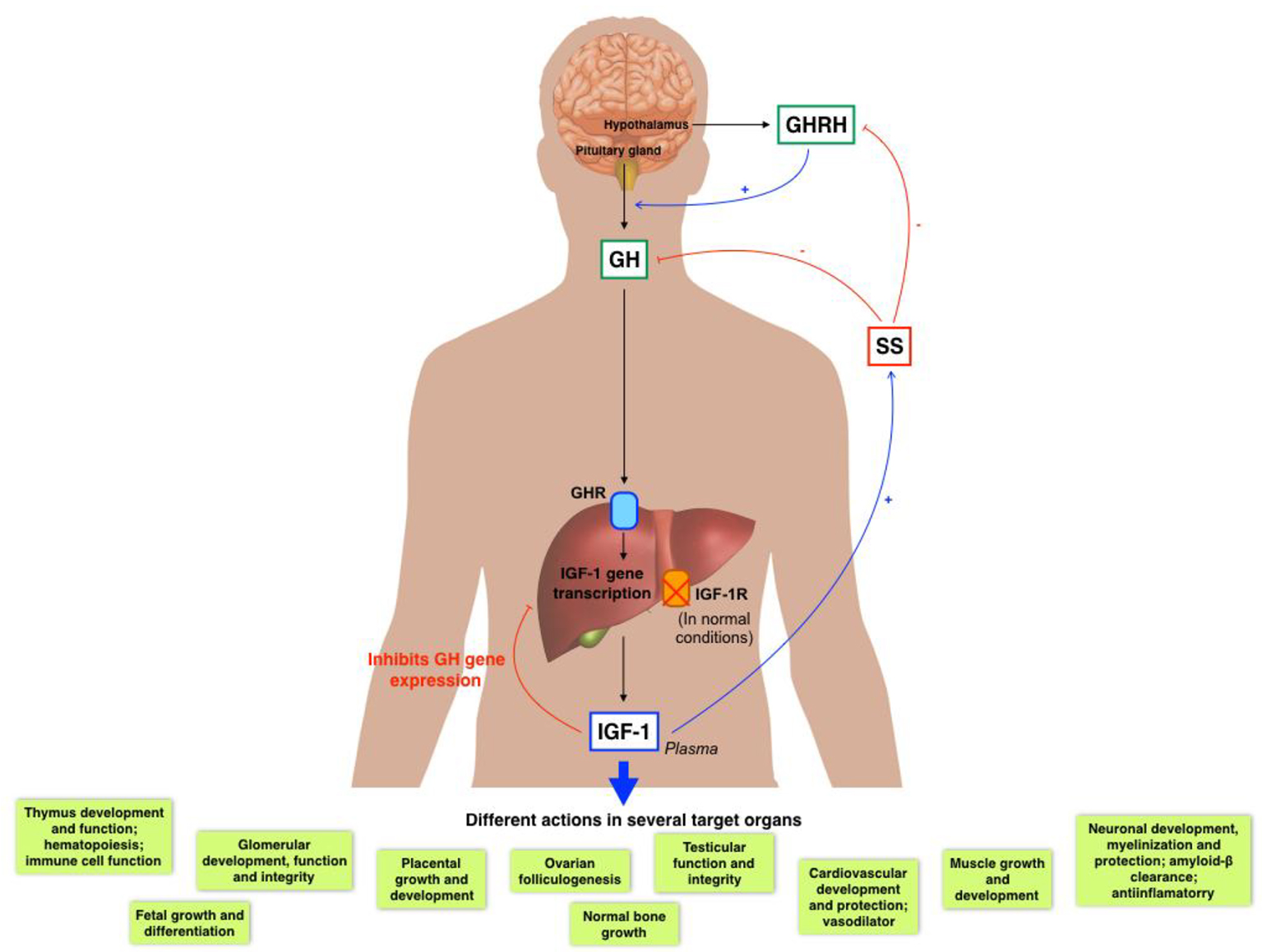 Click for large image | Figure 2. GH/IGF-1 axis and its several actions in diverse organs. |
Circulating IGF-1 can be found in its free form or mainly bound to carrier proteins (IGF binding proteins (IGFBPs)). Because IGF-1 has a wide range of physiological roles, it must be strictly controlled, where IGFBPs play an important role. Until now, there have been identified at least six high affinity IGFBPs. IGFBP-3, which binds 90% of circulating IGF-1, forms a ternary complex consisting of one molecule of IGF-1, one molecule of IGFBP3 and one molecule of the so-called acid labile subunit [31]. GH mainly regulates the IGFBP-3 [32], while IGFBP-1 is mostly regulated by insulin and IGF-1 [33].
In summary, the common functions of IGFBPs are: 1) acting as carrier proteins for circulating IGF-1 and controller of its flow from the vascular space to tissues; 2) increasing IGF-1 half-life and regulating its metabolic clearance [34]; 3) modulating the interaction between IGF-1 and its receptor, and thus indirectly controlling IGF-1 biological activity [29]; 4) modulating IGF-1 in target tissues, inhibiting or activating its specific actions: cell proliferation, differentiation, survival and migration [28, 35-37]; and 5) providing a specific localization pool of IGF-1, because IGFBPs can associate with cell membranes or ECM [38]. Additionally, some IGFBPs may have some biological effects outside IGF-1 signaling pathways, such as apoptosis induction, and proliferation/inhibition in some tumors [37].
Additionally, other nine binding proteins arose as IGFBP-related proteins (IGFBP-rPs), with structural and functional similarities to the IGFBPs but with lesser affinity [34]. Nonetheless, the physiological role of these proteins in the IGF system is not completely defined, but their structural relationship with IGFBPs allows some of these proteins to bind IGF-1, controlling its activity [39, 40].
IGF-1 actions are mediated by its binding to its putative receptor, IGF-1R, a tyrosine kinase. Nonetheless, IGF-1 can also bind to the insulin receptor (with lower affinity), regulating some metabolic functions. Type 1 IGF receptor is a heterotetramer composed of two extracellular α subunits and two transmembrane β subunits. The extracellular α subunits are cysteine-rich regions that confer specificity to ligands, while β subunits have an intracellular part that contains a tyrosine kinase domain, which constitutes the signal transduction mechanism [41]. Tyrosine phosphorylation activates a signaling cascade [42]. IGF-1 has paracrine, endocrine, and autocrine effects on almost every organ, owing this fact to the ubiquitous IGF-1R expression in the organism [30].
The relevance of IGF-1, both in embryological and postnatal states, has been known for years, and its important role in multiple organs has gained recognition more recently. Its wide activities are partly summarized in Figure 3 [22, 43-68].
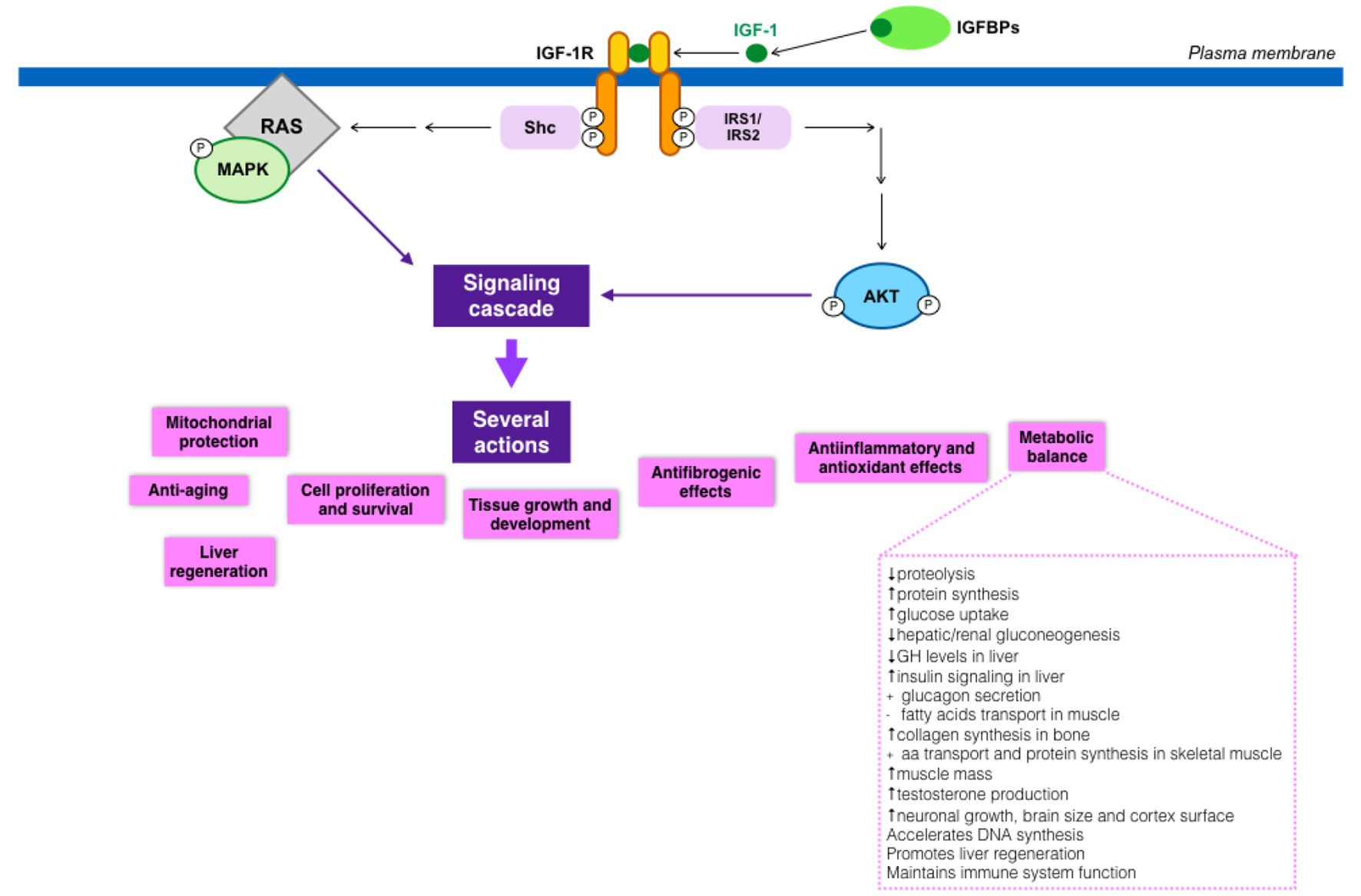 Click for large image | Figure 3. IGF-1 signaling cascade and its implications in metabolism. |
Recent data also support that IGF-1 deficiency is related to insulin resistance, impaired lipid metabolism, oxidative damage and neuro-hormonal axis deregulation [69-71]. Likewise, some studies have also suggested an inverse relationship between IGF-1 circulating levels and the incidence of MetS, with liver steatosis, insulin resistance, hyperlipidemia and abdominal visceral obesity [61, 72-75]. All these results suggest a possible major role of IGF-1 in the development of MetS as well as NASH and NAFLD, which constitute, in many cases, the first stage of metabolic liver damage.
| Physiopathological Pathways Since Early Liver Damage to Decompensated Cirrhosis: Relationship With IGF-1 Deficiency | ▴Top |
The beginning of fibrosis is usually insidious and the progression to cirrhosis can occur in an interval of 15 - 20 years, depending upon different factors - etiology, genetic and environmental aspects [76]. Even though each etiology has its specific pathological feature, a general common pathway can be described from early liver damage all the way to cirrhosis. Hepatic fibrosis is the result of the liver’s response to a repeated injury that can be due to viral infection, MetS with insulin resistance, autoimmune disease, toxins, or alcohol [77]. After an acute injury, the hepatocytes become damaged and an inflammatory response is triggered following HSCs activation, leading to a controlled and coordinated deposition of EMC with parenchymal cells regeneration and replacement of necrotic cells. If the injury persists, this regeneration process fails, and ECM replaces the normal liver parenchyma, through the proliferation and differentiation of HSC to myofibroblasts. Those myofibroblasts secrete different profibrogenic cytokines that finally lead to the synthesis and deposition of fibrillar collagen that forms the ECM. Sinusoidal endothelial cells loose their fenestrations and deposition of ECM increases the resistance to hepatic blood flow. A positive feedback is then established, in which inflammatory and profibrogenic cells stimulate each other, leading to fibrosis, with accumulation of ECM because of the increased synthesis and decreased degradation, as well as the increasing resistance to hepatic blood flow [76].
A common feature that has been described occurring from the early stages of liver damage, central for NAFLD and NASH, is IGF-1 deficiency [78-80]. There are two important aspects of this issue. 1) In both cases (NAFLD and NASH), the insulin resistance plays a major role in the development of liver damage, even though different factors are also implicated (obesity, type 2 diabetes, MetS, and hyperlipidemia, among others) [78]. It is well known that IGF-1 improves insulin sensitivity in vivo, and also that the specific deletion of hepatic IGF-1 results in insulin resistance [81], showing that hepatic IGF-1 regulates systemic insulin sensitivity. 2) As previously mentioned, some recent studies suggest a relationship between IGF-1 deficiency and the risk of developing MetS [61, 82-84], which further would contribute to the appearance of NAFDL and/or NASH.
In conclusion, NAFDL and NASH could be a later manifestation of an IGF-1 deficiency condition, as this deficiency contributes to the presence of different risk factors that lead to the aforementioned diseases. Moreover, IGF-1 deficiency has been described in both entities, so it could represent a common pathway between MetS and hepatic steatosis [78] (Fig. 4).
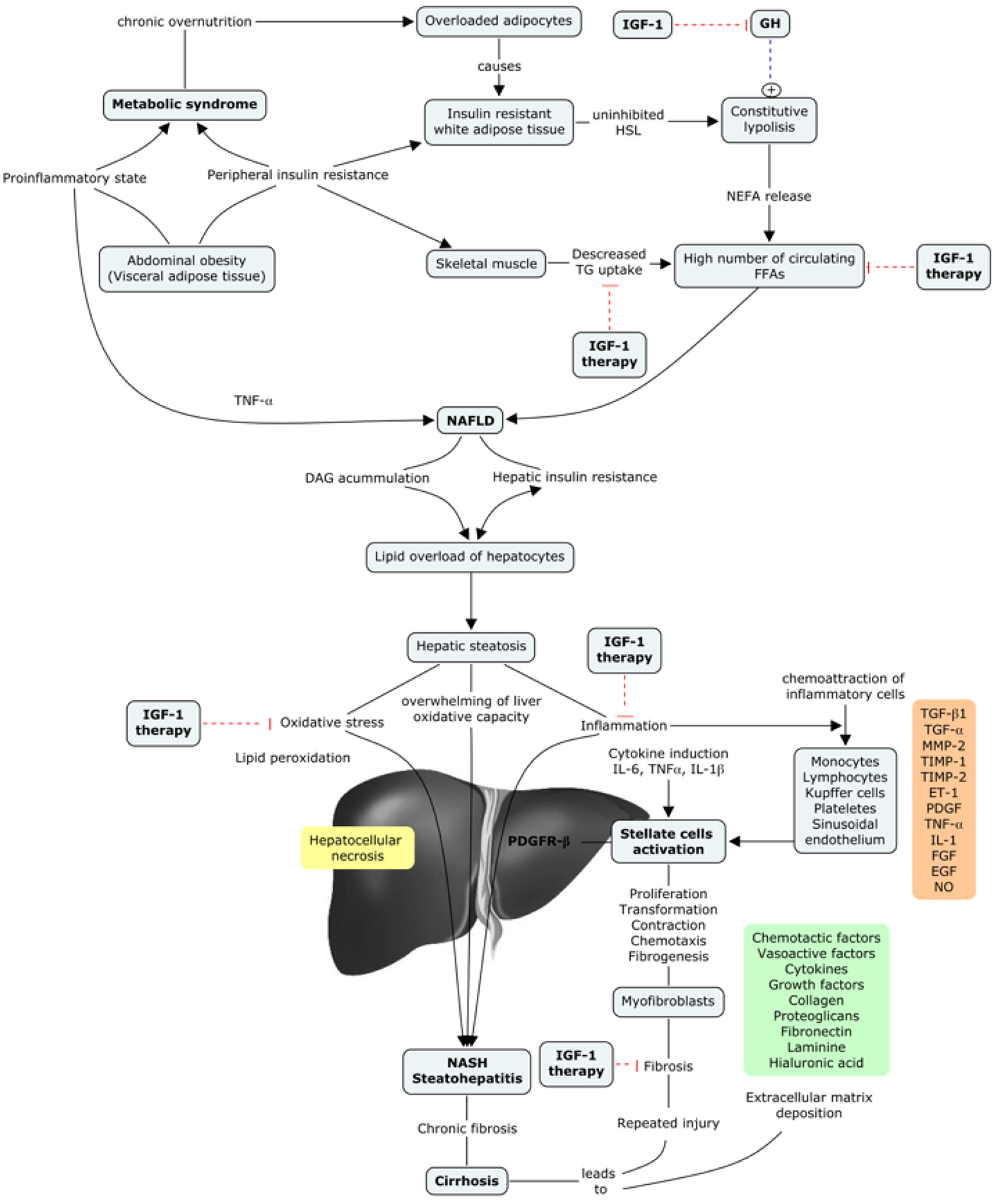 Click for large image | Figure 4. IGF-1 in the progression of metabolic syndrome to liver cirrhosis. |
Numerous different factors take place in this complicated mechanism that can either enhance or ameliorate the activated fibrogenic cascade. A complex interplay between different hepatic cells takes place, leading to the release of reactive oxygen species (ROS) together with fibrogenic and inflammatory mediators, among others. There is growing evidence for the contribution of different immune interactions, chemokines, adipokines, oxidative stress and neuroendocrine factors [76, 77]. Table 2 resumes the inflammatory mediators that are involved in the regulation of fibrogenesis in the liver [85]. These mediators function independently of IGF-1 levels.
Click to view | Table 2. Inflammatory Mediators Implicated in Hepatic Fibrogenesis |
The oxidative stress, mitochondrial dysfunction, and inflammatory cascade, also play an important role in the development and persistence of liver damage of any etiology, leading to fibrosis [76, 77]. Nowadays, it is better understood that all these mechanisms interact with each other promoting fibrosis, and, should they persist activated, the damage will ensue, contributing to cirrhosis stablishment and progression. Nevertheless, the cessation of liver injury has been confirmed to delay and even revert fibrosis at some degree, although this may take several years [77]. In this sense, recent studies focus on the investigation of possible therapeutic targets that could act directly in each profibrogenic pathway, in order to stop this negative stimuli and hence delay or revert fibrosis. However, poor results have been obtained so far.
Despite this, some studies have shown that IGF-1 reduces oxidative stress in the liver, and improves mitochondrial function [86, 87]. Also, it has been described that GH-deficient rats present impaired mitochondrial morphology of the hepatocytes, which improves with IGF-1 administration [88]. As these factors are strongly implicated in the progression of liver damage, it can be a useful target for new therapeutic approaches. In our experience, mitochondria is one of the main intracellular targets of IGF-1, proved in several conditions of IGF-1 deficiency and restored by low doses of this hormone [87].
Moreover, as previously mentioned, it has been recently described that the mere IGF-1 partial deficiency in animals is associated with relevant alterations of the hepatic architecture, as well as an altered expression pattern of genes encoding cytoskeleton proteins, genes related to hepatocyte polarity, cell junctions and ECM proteins, suggesting that IGF-1 deficiency can be strongly implicated since the early stages of liver damage. Additionally, this partial deficiency induced an altered liver expression of genes encoding IGF-1R and proteins involved in acute-phase and inflammation, resulting in hepatic oxidative damage. Also, our group described that cirrhotic animals showed a significant reduction in IGF-1 circulating levels, that IGF-1 treatment restored to normal [89].
Considering all these data, it can be suggested that early liver damage and cirrhosis are IGF-1 deficiency conditions that can be improved with IGF-1 treatment, as the liver expresses IGF-1R under both conditions [11].
| IGF-1 and GH in Cirrhosis | ▴Top |
Liver cirrhosis association with IGF-1 was first described in the late 80s, when it was proposed as a good marker of hepatocellular function. Since then, the idea of liver cirrhosis as a condition of IGF-1 deficiency has been consolidated over the last years.
As previously mentioned, decreased levels of free IGF-1 are observed in patients with chronic liver disease (becoming more severe as the disease progresses) [90-94], despite the normal or elevated GH secretion [91, 95]. This may be due to a decrease in GH receptors on the liver of these patients [96-98], and a progressive reduction of liver synthesis capability. Likewise, IGFBPs production is also modified in cirrhosis, with an increased level of IGFBP-1 and a decreased of IGFBP-3. These changes may play a special role in the bioavailability of IGF-1 in tissues [94, 99, 100], since IGFBP-3 carries up to 80-90% of circulating IGF-1 and maintains plasma concentrations, meanwhile IGFBP-1 sequesters IGF-1, impeding its usage (Fig. 5).
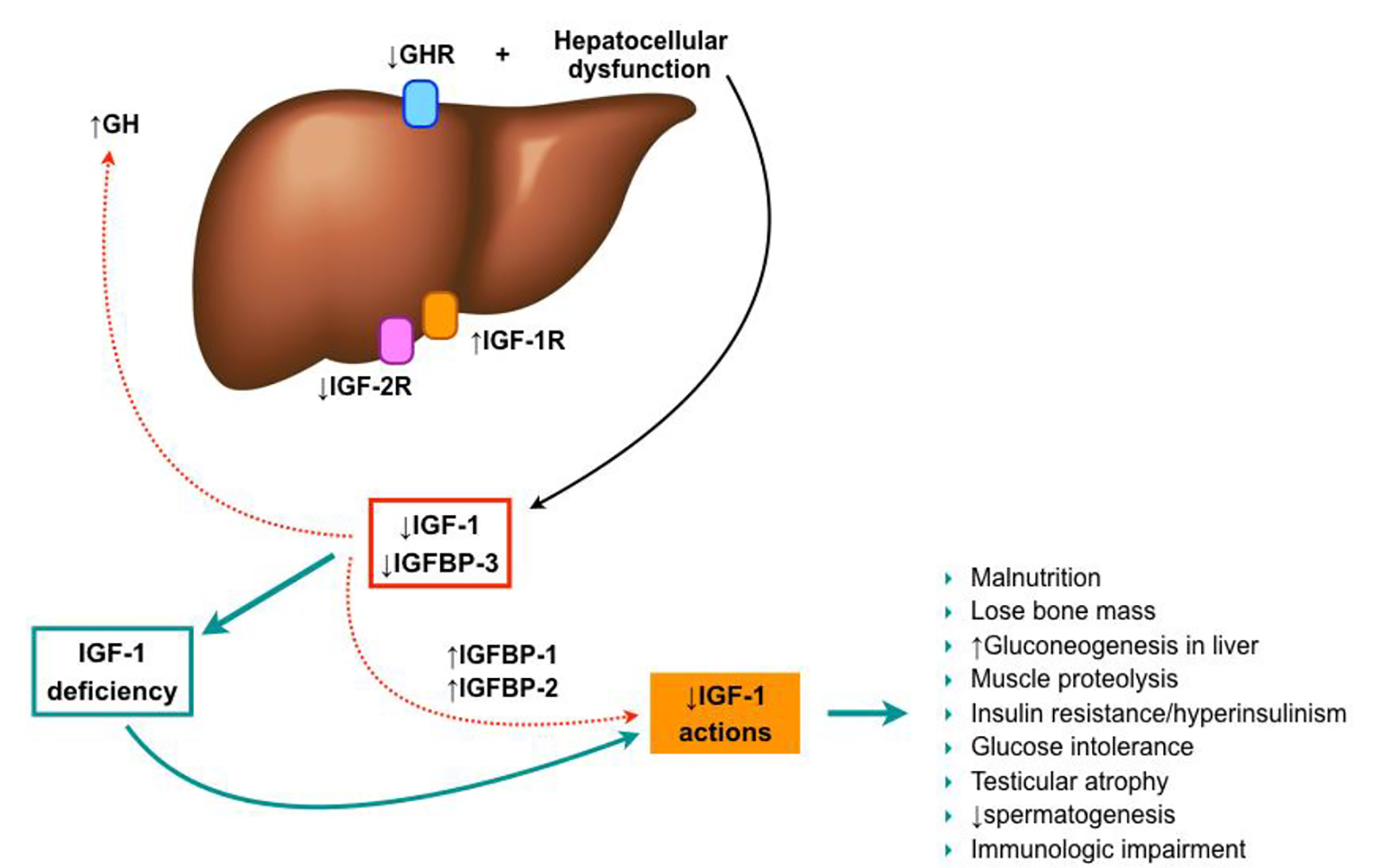 Click for large image | Figure 5. IGF-1 in liver cirrhosis establishment. |
On the other hand, the hepatocytes express few IGF-1 receptors in healthy subjects, so it has been thought that IGF-1 may not affect hepatocyte function directly. However, IGF-1R overexpression in hepatocytes has been described in chronic hepatitis C, chronic hepatitis B, and liver cirrhosis [101-103], when compared with normal livers, suggesting the potential role of IGF-1 in the liver damage under these conditions [104]. Additionally, recent results of an experimental model of IGF-1 partial deficiency induced the expression of IGF-1R in the liver, even though no liver injury is present. So, IGF-1 seems to play an important role in liver homeostasis.
Some other experimental models have shown the physiological importance of GH signaling in the liver. Liver-specific deletion of GH receptor in mice (GHRLD) resulted in a reduction of > 90% of serum IGF-1 levels, contributing to those previously discussed effects [105]. Furthermore, these mice also showed insulin resistance, glucose intolerance, increased fatty acids, decreased triglyceride efflux, severe steatosis, as well as impaired liver regeneration, which proposes that GH may regulate hepatocyte proliferation [65, 106, 107].
Under this scenario, several characteristics of cirrhotic patients can be partially justified by IGF-1 deficiency, such as malnutrition. Glucose production through liver gluconeogenesis is increased in these patients, as well as proteolysis in the muscle. Likewise, they exhibit an increase in insulin and glucose levels partly secondary to insulin resistance, but the exact mechanism is still not well understood. Several causes have been proposed so far, but none of them have been yet confirmed [29, 99, 108]. Additionally, low IGF-1 levels contribute to the loss of bone mass seen in cirrhotic patients [109].
| Results of IGF-1 Therapy | ▴Top |
Results of low doses of recombinant human IGF-1 (rhIGF-1) administration in experimental models of cirrhosis
Following the consolidation of cirrhosis as a condition of IGF-1 deficiency, the replacement therapy with this hormone has been approached in multiple studies in order to test it, given its possible role in the genesis of some cirrhosis complications. Some of theses studies results are discussed below.
Experimental CCl4-induced cirrhosis animals have been treated with rhIGF-1 (20 μg/kg/day for 14 - 21 days) versus placebo, showing the following results: 1) Increased food ingestion, nitrogen balance and the uptake of dietary nitrogen by muscle, leading to increased muscle mass [110, 111]. 2) In vivo and in vitro studies showed recovery of cirrhosis-associated micro-villi atrophy [111-113], as well as carbohydrates and aminoacids intestinal absorption, but no lipid absorption recovery [49, 70, 112, 114, 115]. The intestinal absorption improves as morphologic changes do [115]. 3) Increase of bone density and reduced bone resorption, improving osteopenia, both in compensated and ascitic cirrhosis [116]. 4) Reversion of testicular atrophy as well as histological alteration and improvement of hypofisis-testicular axis. Hemato-testicular barrier is altered from early stages of liver cirrhosis and IGF-1 therapy was able to recover its integrity [117]. Moreover, it produces restoration of the somatoninergic tone, which is reduced in cirrhosis and facilitates the inhibition of GH secretion [118]. 5) Reduced portal pressure, endotoxemia, and bacterial translocation [119], possibly due to the improvement in fibrosis (decreased collagen in liver tissue and histological fibrosis score) [69]. Furthermore, a decrease in collagen synthesis, reducing prolyl-hydroxylase activity and collagen mRNA expression has been also described [50]. Additionally, an improvement in liver function, an increase in albumin and coagulation factor levels, and a reduction in bilirubin levels were found [69]. Some of these changes have been also demonstrated in cirrhosis induced by common bile duct ligation. 6) Regularization of mitochondrial function in the liver [69, 86, 87, 120] and ATPase function, as well as decrease in oxidative stress parameters and free radicals [69]. In particular, a normalization of mitochondrial membrane potential, an increase of ATP production, reducing the intramitochondrial free radical production, as well as a decrease caspase activation and apoptosis have been described [87].
In summary, IGF-1 replacement therapy shows several hepatoprotective, antifibrogenic, anti-inflammatory, and anti-oxidant effects.
Other changes show decrease in lipid peroxidation products and free radicals, decreasing the collagen gene expression in myofibroblasts [121] and prolyl-hydroxylase activity [122], as well as stellate cells activation [123]. Furthermore, the expression of several genes that were altered in CCl4-induced cirrhosis, was normalized after rhIGF-1 replacement therapy [124]. Additionally, the regenerating activity increases with the proliferation of cell nuclear antigen expression, the restoration of GH receptor gene expression, and the stimulation of hepatocyte growth factor production (a potent mitogen and liver protecting agent), as well as the down-regulation of transforming growth factor-β1 (TGF-β1) [125].
Results of IGF-1 gene transfer in experimental models
Since the administration of rhIGF-1 is extremely expensive, it has been proposed that the use of viral vectors encoding IGF-1 can allow sustained expression of the transgene within the cirrhotic liver, helping in treating cirrhosis instead of rhIGF-1 administration [126, 127]. Two studies have been done so far to evaluate this possibility, and Table 3 summarizes the different findings [128]. In the first model, a recombinant simian virus 40 (rSV40) vector encoding for IGF-1 was used to evaluate if the sustained expression of IGF-1 in the liver can protect it against developing cirrhosis after a chronic exposure to CCl4. It was found that rSV40 encoding IGF-1 reduced liver cells damage - showing a decrease in serum transaminases and bilirubin levels - fibrogenesis, and ascites formation as well as improved hypogonadism, showing cytoprotective and antifibrogenic effects [126].
Click to view | Table 3. Factors Up- and Down-Regulated After IGF-1 Gene Transfer in Cirrotic Patients (Modified From Bonefeld and Moller, Liver Int. 2011) |
A second study showed the effects of the administration of rSV40 that encodes for IGF-1 in rats with established cirrhosis. The results showed that this therapy activated different mechanisms for fibrolysis, down-regulated profibrogenic factors, and induced cytoprotective molecules that lead to an improvement of hepatocellular function and reduced fibrosis [127].
Results of rhIGF-1 administration in cirrhotic patients
In human cirrhotic patients, one clinical trial has been conducted, being a pilot, double-blind, randomized, placebo-controlled study in order to evaluate the effects of rhIGF-1 administration in patients with primary biliary cirrhosis or alcohol-related cirrhosis [129]. During 4 months, patients received rhIGF-1 in the following manner: initially 20 μg/kg/day, increasing the dose each week to a maximum dose of 50 μg/kg/day or 100 μg/kg/day for 4 weeks. Even with the limitations of the study, there were three main findings in the patients receiving rhIGF-1: 1) an increase in serum albumin levels, which has never been achieved by any other treatment, 2) a trend towards increased resting energy expenditure and total IGF-1, which can be due to an increased amount of available ATP, because of an improvement of mitochondrial function [87], and 3) an augmented IGF-1/IGFBP3 ratio levels. In this study, IGF-1 was well tolerated and more effective in patients with less nutritional impairment, higher hormone bioavailability rates, and those with alcoholic cirrhosis.
| Other Strategies for Cirrhosis Treatment | ▴Top |
The progression in understanding the pathophysiological mechanisms of cirrhosis has generated new investigations about possible therapeutic drugs that can prevent, delay or reverse fibrosis. Nowadays, the approach to liver fibrosis can be divided into two steps: primary therapy, in order to prevent, delay or even revert fibrosis, comprising in treating the underlying cause (hepatitis B, C, autoimmune hepatitis, alcohol consumption, etc.) [130-134]; secondary therapy, in order to revert fibrosis developing intrinsic antifibrotic drugs that target the fibrogenesis mechanism.
In this scenario, many drugs have been tested so far in experimental animals, but clinical tests of some of them are still pending [76, 135-137]. Moreover, some drugs have shown convincing antifibrotic activity on HSCs in vitro, as well as in animal models of liver fibrosis and even patients in vivo [76, 135-137]. Nevertheless, their long-term safety in cirrhotic patients has not been proven to date.
On the other hand, the reconstitution of functional parenchymal mass in conjunction with fibrosis treatment can lead to a better prognosis [138-140]. In this aspect, hepatocyte transplantation as well as infusion of hepatocyte growth factor, has shown to improve liver function [141, 142]. Moreover, the transplantation of hepatocyte stem cells or progenitor cells promises a better future in the treatment of cirrhotic patients. However, until now, the efficiency of these approaches is still very low, needing further investigations in order to improve the techniques, so they can be applied to patients.
| Conclusions and Future Perspective | ▴Top |
Recently, our knowledge about cirrhosis development and evolution has increased. IGF-1 seems to play an important role in the development and progression of this condition, being a possible marker of the functional reserve of hepatocellular functional capacity [90, 143]. Furthermore, some studies have shown that IGF-1 levels are considered of prognostic value for these patients [143-145]. Additionally, recent studies are overcoming concerns about the implication of IGF-1 deficiency since the early liver damage, even in the absence of liver injury [11].
On the other hand, in the last years, a new therapeutic approach of cirrhosis has been developed, as it no longer considered an irreversible condition. Also, a real effort to delay fibrosis progression has been done, as there is a better understanding of the different mechanisms that contribute to this phenomenon. However, the translation of basic research into improved therapeutics for patients with cirrhosis is still lacking.
Nonetheless, some studies have proved the benefits of IGF-1 treatment in experimental models of steatosis, fibrosis and even cirrhosis, as well as in one clinical trial with cirrhosis patients. Additionally, one study has demonstrated, in an experimental model, the efficacy of using IGF-1 in conjunction with interferon-alpha as a treatment for cirrhosis [89]. In this study, we observed an improvement in liver function biochemistry, hepatic lipid peroxidation and a dramatical reduction of fibrosis, inducing a histological improvement with a recovery of hepatic architecture [89].
In order to continue with the new focus of targeting fibrogenesis pathways, to prevent or delay it, the investigation of IGF-1 as a possible therapeutic agent should seriously be taken into account. In the case of NAFLD and NASH, this therapeutic approach reaches a relevant place, as new studies strongly associated these entities, as well as the MetS, with low levels of IGF-1, making this hormone a perfect candidate to be considered as a possible treatment.
Clinical trials to establish the feasible therapeutic doses of IGF-1 in fibrosis and cirrhosis, as well as its specific contribution in each kind of cirrhosis etiology, could be a relevant research target in the next years.
Acknowledgments
The authors would like to express our gratitude to Jesus Ortiz Urbina, MCs Gabriel Amador Aguirre, Dr. Mariano Garcia-Magarino and Dra. Julieta Rodriguez De Ita for their invaluable help.
Financial Support
No financial support was required for this review article.
Conflicts of Interest
None.
Author Contributions
De la Garza RG contributed to writing the article and reviewing of literature; Morales-Garza LA contributed to article review and advice; Martin-Estal I contributed to reviewing the literature and editing tables and images; and Castilla-Cortazar I contributed to designing the article, reviewing and advice.
Abbreviations
ECM: extracellular matrix; GH: growth hormone; GHRH: growth hormone-releasing hormone; HSCs: hepatic stellate cells; IGFBPs: IGF binding proteins; IGFBP-rPs: IGF related proteins; IGF-1: insulin-like growth factor-1; IGF-1R: IGF-1 receptor; MetS: metabolic syndrome; NASH: non-alcoholic steatohepatitis; NAFLD: non-alcoholic fatty liver disease; rhIGF-1: recombinant human insulin-like growth factor-1; rSV40: recombinant simian virus 40; TGF β1: transforming growth factor β1
| References | ▴Top |
- Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ. 1977;55(4):521-540.
pubmed - Dooley J, Lok A, Burroughs AK HE. Sherlock's Diseases of the Liver and Biliary System. 12th ed. Oxford: Wiley-Blackwell; 2011.
doi - Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838-851.
doi - Bircher J, Benhamou JP, McIntyre N, Rizzetto M RJ. Oxford Textbook of Clinical Hepatology. 2th ed. Oxford University Press; 1999.
- Sherlock S DJ. Diseases of the Liver and Biliary System. 11th ed. Oxford, UK: Blackwell Science; 2002.
- Schiff ER, Sorrell MF ME. Schiff's Diseases of the Liver. 9th ed. Philadelphia: 2003.
- Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet. 2014;383(9930):1749-1761.
doi - Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21(5):1238-1247.
pubmed - Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50(3):604-620.
doi pubmed - Garcia-Pagan JC, Gracia-Sancho J, Bosch J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J Hepatol. 2012;57(2):458-461.
doi pubmed - VJ Lara-Diaz, I Castilla-Cortazar, I Martin-Estal, M Garcia-Magarino, GA Aguirre, JE Puche, MC Sadaba, RG de la Garza, LA Morales UM. IGF-1 deficiency alters 'per se' gene expression of proteins involved in inflammation, cytoskeleton and liver architecture. J Physiol Biochem (Perr Re).
- Garcia-Tsao G, Friedman S, Iredale J, Pinzani M. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51(4):1445-1449.
doi pubmed - Hytiroglou P, Snover DC, Alves V, Balabaud C, Bhathal PS, Bioulac-Sage P, Crawford JM, Dhillon AP, Ferrell L, Guido M, Nakanuma Y, Paradis V, Quaglia A, Theise ND, Thung SN, Tsui WMS, Van Leeuwen DJ. Beyond 'cirrhosis'. Am J Clin Pathol. 2012;137:5-9.
doi pubmed - Quiroga J, Beloqui O, Castilla A. Cirrhosis. In: Prieto J, Rodes J, Schafritz DA. Hepatobiliary Diseases. Berlin: Springer-Verlag; 1992.
- Sarin SK, Dhingra N, Bansal A, Malhotra S, Guptan RC. Dietary and nutritional abnormalities in alcoholic liver disease: a comparison with chronic alcoholics without liver disease. Am J Gastroenterol. 1997;92(5):777-783.
pubmed - Mokdad AA, Lopez AD, Shahraz S, Lozano R, Mokdad AH, Stanaway J, Murray CJ, Naghavi M. Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Med. 2014;12.
doi - Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28(4):629-636.
doi pubmed - Cameron AJ, Shaw JE, Zimmet PZ. The metabolic syndrome: prevalence in worldwide populations. Endocrinol Metab Clin North Am. 2004;33(2):351-375, table of contents.
doi pubmed - Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, et al. The metabolic syndrome. Endocr Rev. 2008;29(7):777-822.
doi pubmed - Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(Suppl 1):S5-10.
pubmed - Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl 1):S99-S112.
doi pubmed - De Ita JR, Castilla-Cortazar I, Aguirre GA, Sanchez-Yago C, Santos-Ruiz MO, Guerra-Menendez L, Martin-Estal I, et al. Altered liver expression of genes involved in lipid and glucose metabolism in mice with partial IGF-1 deficiency: an experimental approach to metabolic syndrome. J Transl Med. 2015;13:326.
doi pubmed - Flier JS, Underhill LH, Le Roith D. Insulin-Like Growth Factors. N Engl J Med. 1997;336:633-640.
doi pubmed - Murphy LJ, Friesen HG. Differential effects of estrogen and growth hormone on uterine and hepatic insulin-like growth factor I gene expression in the ovariectomized hypophysectomized rat. Endocrinology. 1988;122(1):325-332.
doi pubmed - Froesch ER, Schmid C, Schwander J, Zapf J. Actions of insulin-like growth factors. Annu Rev Physiol. 1985;47:443-467.
doi pubmed - Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A. 1999;96(12):7088-7092.
doi pubmed - D'Ercole AJ, Applewhite GT, Underwood LE. Evidence that somatomedin is synthesized by multiple tissues in the fetus. Dev Biol. 1980;75(2):315-328.
doi - Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16(1):3-34.
pubmed - Conchillo M, Prieto J, Quiroga J. [Insulin-like growth factor I (IGF-I) and liver cirrhosis]. Rev Esp Enferm Dig. 2007;99(3):156-164.
doi pubmed - Blomsma MC, de Knegt RJ, Dullaart RP., Jansen PLM. Insulin-like growth factor-I in liver cirrhosis. J. Hepatol. 1997;27:1133-1138.
doi - Lewitt MS, Saunders H, Phuyal JL, Baxter RC. Complex formation by human insulin-like growth factor-binding protein-3 and human acid-labile subunit in growth hormone-deficient rats. Endocrinology. 1994;134(6):2404-2409.
pubmed - Kanety H, Karasik A, Klinger B, Silbergeld A, Laron Z. Long-term treatment of Laron type dwarfs with insulin-like growth factor-1 increases serum insulin-like growth factor-binding protein-3 in the absence of growth hormone activity. Acta Endocrinol (Copenh). 1993;128(2):144-149.
doi - Laron Z, Suikkari AM, Klinger B, Silbergeld A, Pertzelan A, Seppala M, Koivisto VA. Growth hormone and insulin-like growth factor regulate insulin-like growth factor-binding protein-1 in Laron type dwarfism, growth hormone deficiency and constitutional short stature. Acta Endocrinol (Copenh). 1992;127(4):351-358.
doi - Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20(6):761-787.
doi - Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23(6):824-854.
doi pubmed - Yin P, Xu Q, Duan C. Paradoxical actions of endogenous and exogenous insulin-like growth factor-binding protein-5 revealed by RNA interference analysis. J Biol Chem. 2004;279(31):32660-32666.
doi pubmed - Duan C, Xu Q. Roles of insulin-like growth factor (IGF) binding proteins in regulating IGF actions. In: General and Comparative Endocrinology. 2005. page 44-52.
- Oh Y, Nagalla SR, Yamanaka Y, Kim HS, Wilson E, Rosenfeld RG. Synthesis and characterization of insulin-like growth factor-binding protein (IGFBP)-7. Recombinant human mac25 protein specifically binds IGF-I and -II. J Biol Chem. 1996;271(48):30322-30325.
doi pubmed - Burren CP, Wilson EM, Hwa V, Oh Y, Rosenfeld RG. Binding properties and distribution of insulin-like growth factor binding protein-related protein 3 (IGFBP-rP3/NovH), an additional member of the IGFBP Superfamily. J Clin Endocrinol Metab. 1999;84(3):1096-1103.
pubmed - Kim HS, Nagalla SR, Oh Y, Wilson E, Roberts CT, Jr., Rosenfeld RG. Identification of a family of low-affinity insulin-like growth factor binding proteins (IGFBPs): characterization of connective tissue growth factor as a member of the IGFBP superfamily. Proc Natl Acad Sci U S A. 1997;94(24):12981-12986.
doi pubmed - Laron Z. Insulin-like growth factor 1 (IGF-1): a growth hormone. Mol Pathol. 2001;54(5):311-316.
doi pubmed - Werner H, Woloschak M, Stannard B, Shen-Orr Z, Roberts Jr CT LRD. The insulin-like growth factor I receptor: molecular biology, heterogeneity and regulation. In: LeRoith D. Insulin- like growth factors: Molecular and cellular aspects. Boca Raton: 1991. page 17-47.
- Vincent AM, Feldman EL. Control of cell survival by IGF signaling pathways. Growth Horm IGF Res. 2002;12(4):193-197.
doi - Galvan V, Logvinova A, Sperandio S, Ichijo H, Bredesen DE. Type 1 insulin-like growth factor receptor (IGF-IR) signaling inhibits apoptosis signal-regulating kinase 1 (ASK1). J Biol Chem. 2003;278(15):13325-13332.
doi pubmed - Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, et al. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7(12B):2609-2617.
doi pubmed - Locatelli V, Bianchi VE. Effect of GH/IGF-1 on Bone Metabolism and Osteoporsosis. Int J Endocrinol. 2014;2014:235060.
doi pubmed - Jallali N, Ridha H, Thrasivoulou C, Butler P, Cowen T. Modulation of intracellular reactive oxygen species level in chondrocytes by IGF-1, FGF, and TGF-beta1. Connect Tissue Res. 2007;48(3):149-158.
doi pubmed - Garcia-Fernandez M, Castilla-Cortazar I, Diaz-Sanchez M, Diez Caballero F, Castilla A, Diaz Casares A, Varela-Nieto I, et al. Effect of IGF-I on total serum antioxidant status in cirrhotic rats. J Physiol Biochem. 2003;59(2):145-146.
doi pubmed - Garcia-Fernandez M, Castilla-Cortazar I, Diaz-Sanchez M, Navarro I, Puche JE, Castilla A, Casares AD, et al. Antioxidant effects of insulin-like growth factor-I (IGF-I) in rats with advanced liver cirrhosis. BMC Gastroenterol. 2005;5:7.
doi pubmed - Muguerza B, Castilla-Cortazar I, Garcia M, Quiroga J, Santidrian S, Prieto J. Antifibrogenic effect in vivo of low doses of insulin-like growth factor-I in cirrhotic rats. Biochim Biophys Acta. 2001;1536(2-3):185-195.
doi - Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci. 2012;67(6):626-639.
doi pubmed - Puche JE, Garcia-Fernandez M, Muntane J, Rioja J, Gonzalez-Baron S, Castilla Cortazar I. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology. 2008;149(5):2620-2627.
doi pubmed - Laager R, Ninnis R, Keller U. Comparison of the effects of recombinant human insulin-like growth factor-I and insulin on glucose and leucine kinetics in humans. J Clin Invest. 1993;92(4):1903-1909.
doi pubmed - Pratipanawatr T, Pratipanawatr W, Rosen C, Berria R, Bajaj M, Cusi K, Mandarino L, et al. Effect of IGF-I on FFA and glucose metabolism in control and type 2 diabetic subjects. Am J Physiol Endocrinol Metab. 2002;282(6):E1360-1368.
doi pubmed - Moxley RT, 3rd, Arner P, Moss A, Skottner A, Fox M, James D, Livingston JN. Acute effects of insulin-like growth factor I and insulin on glucose metabolism in vivo. Am J Physiol. 1990;259(4 Pt 1):E561-567.
pubmed - Jacob R, Barrett E, Plewe G, Fagin KD, Sherwin RS. Acute effects of insulin-like growth factor I on glucose and amino acid metabolism in the awake fasted rat. Comparison with insulin. J Clin Invest. 1989;83(5):1717-1723.
doi pubmed - Pennisi P, Gavrilova O, Setser-Portas J, Jou W, Santopietro S, Clemmons D, Yakar S, et al. Recombinant human insulin-like growth factor-I treatment inhibits gluconeogenesis in a transgenic mouse model of type 2 diabetes mellitus. Endocrinology. 2006;147(6):2619-2630.
doi pubmed - Fernandez AM, Kim JK, Yakar S, Dupont J, Hernandez-Sanchez C, Castle AL, Filmore J, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15(15):1926-1934.
doi pubmed - Mauras N, O'Brien KO, Welch S, Rini A, Helgeson K, Vieira NE, Yergey AL. Insulin-like growth factor I and growth hormone (GH) treatment in GH-deficient humans: differential effects on protein, glucose, lipid, and calcium metabolism. 2000.
- Heron-Milhavet L, Haluzik M, Yakar S, Gavrilova O, Pack S, Jou WC, Ibrahimi A, et al. Muscle-specific overexpression of CD36 reverses the insulin resistance and diabetes of MKR mice. Endocrinology. 2004;145(10):4667-4676.
doi pubmed - Aguirre GA, De Ita JR, de la Garza RG, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med. 2016;14:3.
doi pubmed - Kudo Y, Iwashita M, Iguchi T, Takeda Y. The regulation of L-proline transport by insulin-like growth factor-I in human osteoblast-like SaOS-2 cells. Pflugers Arch. 1996;432(3):419-425.
doi pubmed - Thoren MC, Wivall-Helleryd IL, Blum WF, Hall KE. Effects of repeated subcutaneous administration of recombinant human insulin-like growth factor I in adults with growth hormone deficiency. Eur J Endocrinol. 1994;131(1):33-40.
doi pubmed - Desbois-Mouthon C, Wendum D, Cadoret A, Rey C, Leneuve P, Blaise A, Housset C, et al. Hepatocyte proliferation during liver regeneration is impaired in mice with liver-specific IGF-1R knockout. FASEB J. 2006;20(6):773-775.
doi - Pennisi PA, Kopchick JJ, Thorgeirsson S, LeRoith D, Yakar S. Role of growth hormone (GH) in liver regeneration. Endocrinology. 2004;145(10):4748-4755.
doi pubmed - Walsh PT, Smith LM, O'Connor R. Insulin-like growth factor-1 activates Akt and Jun N-terminal kinases (JNKs) in promoting the survival of T lymphocytes. Immunology. 2002;107(4):461-471.
doi pubmed - Kelley KW, Weigent DA, Kooijman R. Protein hormones and immunity. Brain Behav Immun. 2007;21(4):384-392.
doi pubmed - Tu W, Cheung PT, Lau YL. Insulin-like growth factor 1 promotes cord blood T cell maturation and inhibits its spontaneous and phytohemagglutinin-induced apoptosis through different mechanisms. J Immunol. 2000;165(3):1331-1336.
doi pubmed - Castilla-Cortazar I, Garcia M, Muguerza B, Quiroga J, Perez R, Santidrian S, Prieto J. Hepatoprotective effects of insulin-like growth factor I in rats with carbon tetrachloride-induced cirrhosis. Gastroenterology. 1997;113(5):1682-1691.
doi pubmed - Garcia-Fernandez M, Delgado G, Puche JE, Gonzalez-Baron S, Castilla Cortazar I. Low doses of insulin-like growth factor I improve insulin resistance, lipid metabolism, and oxidative damage in aging rats. Endocrinology. 2008;149(5):2433-2442.
doi pubmed - Liu W, Li WB, Chen ZJ, Rong ZH, Chang LW. [Protective effects of IGF-1 on cortical nerve cells of neonatal rats under oxidative stress]. Zhongguo Dang Dai Er Ke Za Zhi. 2014;16(2):203-207.
pubmed - Malik J, Stulc T, Wichterle D, Melenovsky V, Chytilova E, Lacinova Z, Marek J, et al. Hyperlipidemia is associated with altered levels of insulin-like growth factor-I. Physiol Res. 2008;57(6):919-925.
pubmed - Mallea-Gil MS, Ballarino MC, Spiraquis A, Iriarte M, Kura M, Gimenez S, Oneto A, et al. IGF-1 levels in different stages of liver steatosis and its association with metabolic syndrome. Acta Gastroenterol Latinoam. 2012;42(1):20-26.
pubmed - Franco C, Bengtsson BA, Johannsson G. The GH/IGF-1 Axis in Obesity: Physiological and Pathological Aspects. Metab Syndr Relat Disord. 2006;4(1):51-56.
doi pubmed - Moses AC. Insulin resistance and type 2 diabetes mellitus: is there a therapeutic role for IGF-1? Endocr Dev. 2005;9:121-134.
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209-218.
doi pubmed - Elpek GO. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J Gastroenterol. 2014;20(23):7260-7276.
doi pubmed - Takahashi Y. Essential roles of growth hormone (GH) and insulin-like growth factor-I (IGF-I) in the liver. Endocr J. 2012;59(11):955-962.
doi pubmed - Arturi F, Succurro E, Procopio C, Pedace E, Mannino GC, Lugara M, Procopio T, et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab. 2011;96(10):E1640-1644.
doi pubmed - Volzke H, Nauck M, Rettig R, Dorr M, Higham C, Brabant G, Wallaschofski H. Association between hepatic steatosis and serum IGF1 and IGFBP-3 levels in a population-based sample. Eur J Endocrinol. 2009;161(5):705-713.
doi pubmed - Yakar S, Liu JL, Fernandez AM, Wu Y, Schally AV, Frystyk J, Chernausek SD, et al. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes. 2001;50(5):1110-1118.
doi pubmed - Lam CS, Chen MH, Lacey SM, Yang Q, Sullivan LM, Xanthakis V, Safa R, et al. Circulating insulin-like growth factor-1 and its binding protein-3: metabolic and genetic correlates in the community. Arterioscler Thromb Vasc Biol. 2010;30(7):1479-1484.
doi pubmed - Akanji AO, Smith RJ. The insulin-like growth factor system, metabolic syndrome, and cardiovascular disease risk. Metab Syndr Relat Disord. 2012;10(1):3-13.
doi pubmed - Sierra-Johnson J, Romero-Corral A, Somers VK, Lopez-Jimenez F, Malarstig A, Brismar K, Hamsten A, et al. IGF-I/IGFBP-3 ratio: a mechanistic insight into the metabolic syndrome. Clin Sci (Lond). 2009;116(6):507-512.
doi pubmed - Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61(3):1066-1079.
doi pubmed - Hao CN, Geng YJ, Li F, Yang T, Su DF, Duan JL, Li Y. Insulin-like growth factor-1 receptor activation prevents hydrogen peroxide-induced oxidative stress, mitochondrial dysfunction and apoptosis. Apoptosis. 2011;16(11):1118-1127.
doi pubmed - Perez R, Garcia-Fernandez M, Diaz-Sanchez M, Puche JE, Delgado G, Conchillo M, Muntane J, et al. Mitochondrial protection by low doses of insulin-like growth factor- I in experimental cirrhosis. World J Gastroenterol. 2008;14(17):2731-2739.
doi pubmed - Nishizawa H, Takahashi M, Fukuoka H, Iguchi G, Kitazawa R, Takahashi Y. GH-independent IGF-I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem Biophys Res Commun. 2012;423(2):295-300.
doi pubmed - Tutau F, Rodriguez-Ortigosa C, Puche JE, Juanarena N, Monreal I, Garcia Fernandez M, Clavijo E, et al. Enhanced actions of insulin-like growth factor-I and interferon-alpha co-administration in experimental cirrhosis. Liver Int. 2009;29(1):37-46.
doi pubmed - Caufriez A, Reding P, Urbain D, Golstein J, Copinschi G. Insulin-like growth factor I: a good indicator of functional hepatocellular capacity in alcoholic liver cirrhosis. J Endocrinol Invest. 1991;14(4):317-321.
doi pubmed - Cuneo RC, Hickman PE, Wallace JD, Teh BT, Ward G, Veldhuis JD, Waters MJ. Altered endogenous growth hormone secretory kinetics and diurnal GH-binding protein profiles in adults with chronic liver disease. Clin Endocrinol (Oxf). 1995;43(3):265-275.
doi - Donaghy A, Ross R, Wicks C, Hughes SC, Holly J, Gimson A, Williams R. Growth hormone therapy in patients with cirrhosis: a pilot study of efficacy and safety. Gastroenterology. 1997;113(5):1617-1622.
doi pubmed - Assy N, Hochberg Z, Amit T, Shen-Orr Z, Enat R, Baruch Y. Growth hormone-stimulated insulin-like growth factor (IGF) I and IGF-binding protein-3 in liver cirrhosis. J Hepatol. 1997;27(5):796-802.
doi - Scharf JG, Schmitz F, Frystyk J, Skjaerbaek C, Moesus H, Blum WF, Ramadori G, et al. Insulin-like growth factor-I serum concentrations and patterns of insulin-like growth factor binding proteins in patients with chronic liver disease. J Hepatol. 1996;25(5):689-699.
doi - Clemmons DR, Klibanski A, Underwood LE, McArthur JW, Ridgway EC, Beitins IZ, Van Wyk JJ. Reduction of plasma immunoreactive somatomedin C during fasting in humans. J Clin Endocrinol Metab. 1981;53(6):1247-1250.
doi pubmed - Chang TC, Lin JJ, Yu SC, Chang TJ. Absence of growth-hormone receptor in hepatocellular carcinoma and cirrhotic liver. Hepatology. 1990;11(1):123-126.
doi pubmed - Donaghy AJ, Delhanty PJ, Ho KK, Williams R, Baxter RC. Regulation of the growth hormone receptor/binding protein, insulin-like growth factor ternary complex system in human cirrhosis. J Hepatol. 2002;36(6):751-758.
doi - Shen XY, Holt RI, Miell JP, Justice S, Portmann B, Postel-Vinay MC, Ross RJ. Cirrhotic liver expresses low levels of the full-length and truncated growth hormone receptors. J Clin Endocrinol Metab. 1998;83(7):2532-2538.
doi - Shmueli E, Miell JP, Stewart M, Alberti KG, Record CO. High insulin-like growth factor binding protein 1 levels in cirrhosis: link with insulin resistance. Hepatology. 1996;24(1):127-133.
doi pubmed - Donaghy A, Ross R, Gimson A, Hughes SC, Holly J, Williams R. Growth hormone, insulinlike growth factor-1, and insulinlike growth factor binding proteins 1 and 3 in chronic liver disease. Hepatology. 1995;21(3):680-688.
pubmed - Stefano JT, Correa-Giannella ML, Ribeiro CM, Alves VA, Massarollo PC, Machado MC, Giannella-Neto D. Increased hepatic expression of insulin-like growth factor-I receptor in chronic hepatitis C. World J Gastroenterol. 2006;12(24):3821-3828.
doi pubmed - Kim SO, Park JG, Lee YI. Increased expression of the insulin-like growth factor I (IGF-I) receptor gene in hepatocellular carcinoma cell lines: implications of IGF-I receptor gene activation by hepatitis B virus X gene product. Cancer Res. 1996;56(16):3831-3836.
pubmed - Tao X, Shen D, Ren H, Zhang X, Zhang D, Ye J, Gu B. Hepatitis B virus X protein activates expression of IGF-IR and VEGF in hepatocellular carcinoma cells. Zhonghua Gan Zang Bing Za Zhi. 2000;8(3):161-163.
pubmed - Aleem E, Nehrbass D, Klimek F, Mayer D, Bannasch P. Upregulation of the insulin receptor and type I insulin-like growth factor receptor are early events in hepatocarcinogenesis. Toxicol Pathol. 2011;39(3):524-543.
doi pubmed - Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, Kopchick JJ, et al. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284(30):19937-19944.
doi pubmed - Ekberg S, Luther M, Nakamura T, Jansson JO. Growth hormone promotes early initiation of hepatocyte growth factor gene expression in the liver of hypophysectomized rats after partial hepatectomy. J Endocrinol. 1992;135(1):59-67.
doi pubmed - Krupczak-Hollis K, Wang X, Dennewitz MB, Costa RH. Growth hormone stimulates proliferation of old-aged regenerating liver through forkhead box m1b. Hepatology. 2003;38(6):1552-1562.
doi pubmed - Petersen KF, Jacob R, West AB, Sherwin RS, Shulman GI. Effects of insulin-like growth factor I on glucose metabolism in rats with liver cirrhosis. Am J Physiol. 1997;273(6 Pt 1):E1189-1193.
pubmed - Gallego-Rojo FJ, Gonzalez-Calvin JL, Munoz-Torres M, Mundi JL, Fernandez-Perez R, Rodrigo-Moreno D. Bone mineral density, serum insulin-like growth factor I, and bone turnover markers in viral cirrhosis. Hepatology. 1998;28(3):695-699.
doi pubmed - Picardi A, de Oliveira AC, Muguerza B, Tosar A, Quiroga J, Castilla-Cortazar I, Santidrian S, et al. Low doses of insulin-like growth factor-I improve nitrogen retention and food efficiency in rats with early cirrhosis. J Hepatol. 1997;26(1):191-202.
doi - Castilla-Cortazar I, Prieto J, Urdaneta E, Pascual M, Nunez M, Zudaire E, Garcia M, et al. Impaired intestinal sugar transport in cirrhotic rats: correction by low doses of insulin-like growth factor I. Gastroenterology. 1997;113(4):1180-1187.
doi pubmed - Castilla-Cortazar I, Picardi A, Tosar A, Ainzua J, Urdaneta E, Garcia M, Pascual M, et al. Effect of insulin-like growth factor I on in vivo intestinal absorption of D-galactose in cirrhotic rats. Am J Physiol. 1999;276(1 Pt 1):G37-42.
pubmed - Castilla-Cortazar I, Pascual M, Urdaneta E, Pardo J, Puche JE, Vivas B, Diaz-Casares A, et al. Jejunal microvilli atrophy and reduced nutrient transport in rats with advanced liver cirrhosis: improvement by Insulin-like Growth Factor I. BMC Gastroenterol. 2004;4:12.
doi pubmed - Pascual M, Castilla-Cortazar I, Urdaneta E, Quiroga J, Garcia M, Picardi A, Prieto J. Altered intestinal transport of amino acids in cirrhotic rats: the effect of insulin-like growth factor-I. Am J Physiol Gastrointest Liver Physiol. 2000;279(2):G319-324.
pubmed - Perez R, Castilla-Cortazar I, Nunez M, Prado A, Mirpuri E, Garcia M, Gonzalez Baron S, et al. IGF-I does not improve fat malabsorption in cirrhotic rats. J Physiol Biochem. 2001;57(1):59-60.
doi - Cemborain A, Castilla-Cortazar I, Garcia M, Quiroga J, Muguerza B, Picardi A, Santidrian S, et al. Osteopenia in rats with liver cirrhosis: beneficial effects of IGF-I treatment. J Hepatol. 1998;28(1):122-131.
doi - Castilla-Cortazar I, Garcia M, Quiroga J, Diez N, Diez-Caballero F, Calvo A, Diaz M, et al. Insulin-like growth factor-I reverts testicular atrophy in rats with advanced cirrhosis. Hepatology. 2000;31(3):592-600.
doi pubmed - Castilla-Cortazar I, Aliaga-Montilla MA, Salvador J, Garcia M, Delgado G, Gonzalez-Baron S, Quiroga J, et al. Insulin-like growth factor-I restores the reduced somatostatinergic tone controlling growth hormone secretion in cirrhotic rats. Liver. 2001;21(6):405-409.
doi pubmed - Lorenzo-Zuniga V, Rodriguez-Ortigosa CM, Bartoli R, Martinez-Chantar ML, Martinez-Peralta L, Pardo A, Ojanguren I, et al. Insulin-like growth factor I improves intestinal barrier function in cirrhotic rats. Gut. 2006;55(9):1306-1312.
doi pubmed - Garcia-Fernandez M, Sierra I, Puche JE, Guerra L, Castilla-Cortazar I. Liver mitochondrial dysfunction is reverted by insulin-like growth factor II (IGF-II) in aging rats. J Transl Med. 2011;9:123.
doi pubmed - Bedossa P, Houglum K, Trautwein C, Holstege A, Chojkier M. Stimulation of collagen alpha 1(I) gene expression is associated with lipid peroxidation in hepatocellular injury: a link to tissue fibrosis? Hepatology. 1994;19(5):1262-1271.
pubmed - Yamada S, Yamada M, Murawaki Y, Hirayama C. Increase in lipoperoxides and prolyl hydroxylase activity in rat liver following chronic ethanol feeding. Biochem Pharmacol. 1990;40(5):1015-1019.
doi - AJ MC. Complications of chronic liver disease. St. Louis: Mosby Year Book; 1992.
- Mirpuri E, Garcia-Trevijano ER, Castilla-Cortazar I, Berasain C, Quiroga J, Rodriguez-Ortigosa C, Mato JM, et al. Altered liver gene expression in CCl4-cirrhotic rats is partially normalized by insulin-like growth factor-I. Int J Biochem Cell Biol. 2002;34(3):242-252.
doi - Skrtic S, Wallenius K, Gressner AM, Jansson JO. Insulin-like growth factor signaling pathways in rat hepatic stellate cells: importance for deoxyribonucleic acid synthesis and hepatocyte growth factor production. Endocrinology. 1999;140(12):5729-5735.
doi - Vera M, Sobrevals L, Zaratiegui M, Martinez L, Palencia B, Rodriguez CM, Prieto J, et al. Liver transduction with a simian virus 40 vector encoding insulin-like growth factor I reduces hepatic damage and the development of liver cirrhosis. Gene Ther. 2007;14(3):203-210.
doi pubmed - Sobrevals L, Rodriguez C, Romero-Trevejo JL, Gondi G, Monreal I, Paneda A, Juanarena N, et al. Insulin-like growth factor I gene transfer to cirrhotic liver induces fibrolysis and reduces fibrogenesis leading to cirrhosis reversion in rats. Hepatology. 2010;51(3):912-921.
pubmed - Bonefeld K, Moller S. Insulin-like growth factor-I and the liver. Liver Int. 2011;31(7):911-919.
doi pubmed - Conchillo M, de Knegt RJ, Payeras M, Quiroga J, Sangro B, Herrero JI, Castilla-Cortazar I, et al. Insulin-like growth factor I (IGF-I) replacement therapy increases albumin concentration in liver cirrhosis: results of a pilot randomized controlled clinical trial. J Hepatol. 2005;43(4):630-636.
doi pubmed - Arthur MJ. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122(5):1525-1528.
doi pubmed - Dixon JB, Bhathal PS, Hughes NR, O'Brien PE. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology. 2004;39(6):1647-1654.
doi pubmed - Poynard T, McHutchison J, Manns M, Trepo C, Lindsay K, Goodman Z, Ling MH, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122(5):1303-1313.
doi pubmed - Dienstag JL, Goldin RD, Heathcote EJ, Hann HW, Woessner M, Stephenson SL, Gardner S, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124(1):105-117.
doi pubmed - Dufour JF, DeLellis R, Kaplan MM. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann Intern Med. 1997;127(11):981-985.
doi pubmed - Friedman SL. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1(2):98-105.
doi pubmed - Pinzani M, Rombouts K, Colagrande S. Fibrosis in chronic liver diseases: diagnosis and management. J Hepatol. 2005;42 Suppl(1):S22-36.
- Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3(2):95-107.
doi - Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39(6):1477-1487.
doi pubmed - Gupta S, Chowdhury JR. Therapeutic potential of hepatocyte transplantation. Semin Cell Dev Biol. 2002;13(6):439-446.
doi pubmed - Strom S, Fisher R. Hepatocyte transplantation: new possibilities for therapy. Gastroenterology. 2003;124(2):568-571.
doi pubmed - Kobayashi N, Ito M, Nakamura J, Cai J, Gao C, Hammel JM, Fox IJ. Hepatocyte transplantation in rats with decompensated cirrhosis. Hepatology. 2000;31(4):851-857.
doi pubmed - Ahmad TA, Eguchi S, Yanaga K, Miyamoto S, Kamohara Y, Fujioka H, Furui J, et al. Role of intrasplenic hepatocyte transplantation in improving survival and liver regeneration after hepatic resection in cirrhotic rats. Cell Transplant. 2002;11(5):399-402.
pubmed - Caregaro L, Alberino F, Amodio P, Merkel C, Angeli P, Plebani M, Bolognesi M, et al. Nutritional and prognostic significance of insulin-like growth factor 1 in patients with liver cirrhosis. Nutrition. 1997;13(3):185-190.
doi - Min J, Yu H, Yan H, He L, Liu H, Zhao C. [The growth hormone and insulin-like growth factors axis in liver failure patients]. Zhonghua Gan Zang Bing Za Zhi. 2001;9(Suppl):76-78.
pubmed - Moller S, Becker PU, Juul A, Skakkebaek NE, Christensen E. [Prognostic value of insulin-like growth factor I-IGF-I - and its binding protein IGFBP-3 in alcoholic liver disease]. Ugeskr Laeger. 1997;159(30):4636-4640.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited..
Journal of Clinical Medicine Research is published by Elmer Press Inc.