

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 8, Number 2, February 2016, pages 63-75
Novel Drugs and Combination Therapies for the Treatment of Metastatic Melanoma
Adarsh Vennepureddya, c, Nishitha Thumallapallya, Vijeyaluxmy Motilal Nehrua, Jean-Paul Atallahb, Terenig Terjanianb
aDepartment of Medicine, Staten Island University Hospital, Staten Island, NY 10305, USA.
bDivision of Hematolgy and Oncology, Staten Island University Hospital, Staten Island, NY 10305, USA
cCorresponding Author: Adarsh Vennepureddy, Staten Island University Hospital, 475 Seaview Avenue, Staten Island, NY 10305, USA
Manuscript accepted for publication December 07, 2015
Short title: Drugs for Metastatic Melanoma
doi: http://dx.doi.org/10.14740/jocmr2424w
- Abstract
- Introduction
- Mechanism of Action of Immunotherapy
- Ipilimumab
- Pembrolizumab
- Nivolumab
- AEs of Immunotherapy
- Management of AEs
- Mechanism of Action of Targeted Therapy
- Vemurafenib
- Dabrafenib
- Trametinib
- Combination Therapy
- Combined BRAF and MEK Inhibitor
- Conclusion and Approach to Therapy
- References
| Abstract | ▴Top |
Metastatic melanoma (MM) still remains as one of the most worrisome cancer known to mankind. In last two decades, treatment of melanoma took a dramatic turn with the discovery of targeted therapy which targets the mutations in mitogen-activated protein kinase (MAPK) pathway and immune checkpoint inhibitors. These new findings have led to emergence of many novel drugs that have been approved by FDA. Targeted therapy drugs such as vemurafenib, trametinib and dabrafenib target the MAPK pathway whereas immunotherapies such as ipilimumab, nivolumab and pembrolizumab block immune checkpoint receptors on T lymphocytes. All these drugs have shown to improve the overall survival in MM. Despite these recent discoveries, treatment of MM remains challenging because of rapid development of resistance to targeted therapy. This review will discuss recently approved drugs and their adverse effects and also shed light on combination therapy in treatment of melanoma.
Keywords: Metastatic melanoma; Targeted therapy; Immunotherapy; Combination therapy
| Introduction | ▴Top |
Melanoma is the leading cause of death from skin disease. It has been reported as fifth and seventh most common cancer in USA in men and women respectively [1]. According to the National Cancer Institute (NCI), an estimated 73,870 new cases of melanoma will be diagnosed in the United States in 2015, and about 9,180 people would have died from the disease in 2014. The incidence of melanoma additionally varies by ethnic group. It accounts for 1 (per 100,000) in black people, four in Hispanics, and 25 in non-Hispanic whites annually [1]. The 10-year overall survival (OS) rate for advanced melanoma is about 10-15% and in the elderly (age > 70), regardless of their disease stage, the survival rate drops dramatically [2]. Majority of melanoma lesions are diagnosed early and are mostly excised and curable. But the real challenge lies in treating advanced melanoma. Treatment of melanoma depends on the stage on presentation. As per NCI, excision is treatment of choice for stage 0 melanoma. Stage II, III and resectable melanoma are managed with excision and lymph node resection if involved and unresectable stage III and IV melanoma are treated with help of chemotherapy, targeted therapy and immunotherapy [3].
Treatment of metastatic melanoma (MM) has changed drastically over the last decade. Historically, melanomas were considered as a single disease entity and treatment options included radiation therapy/surgery or chemotherapy with dacarbazine, an alkylating agent. However response to chemotherapy was nominal and durable remission rarely occurred. Encountered with un-satisfactory results with traditional chemotherapy, focus was shifted on learning in depth pathogenesis of melanoma at cellular and molecular level. With extended knowledge in molecular medicine, melanoma has been reclassified as a highly complex heterogenous disease comprising of several subpopulation of tumor cells. Number of gene mutations and aberrant cell signaling pathways have been recognized which led to development of targeted therapy and immunotherapy drugs. Although these new drugs show dramatic increase in overall response rate and extended survival, treatment of advanced melanoma still remains a challenge [4-6].
In recent years, four different classes of novel drugs were approved revolutionizing the care of advanced melanoma. These include immunotherapy (anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4) monoclonal antibodies; anti-programmed cell death-1 protein (PD-1) monoclonal antibodies), targeted therapy like BRAF inhibitors and MEK inhibitors.
In this review, we discussed the development and current status of the expanding landscape of melanoma treatment.
| Mechanism of Action of Immunotherapy | ▴Top |
Under normal physiologic conditions, the immune checkpoints serve to restrain immune responses against self-antigens, thereby preventing unwanted autoimmunity. However, these inhibitory pathways are up-regulated in many cancers, and immune checkpoints play critical roles in cancer-associated immune suppression and immune evasion [7].
Anti-CTLA-4 antibodies
The primary effector cells of the adaptive immune response against cancer are the T lymphocytes which include both T helper cells and cytotoxic T lymphocytes. Cytotoxic T lymphocytes have direct tumor killing ability and T helper cells play a pivotal role in propagating anti-tumor response. T-cell activation requires two sequential signals. In a first step, antigens presented in context with the major histocompatibility complex (MHC) I or II on specialized antigen-presenting cells (APCs) bind with T-cell receptors (TCRs). The second step involves translation of TCR stimulation into T-cell activation and requires a co-stimulatory signal, achieved when B7 molecules on the APC surface bind with CD28 receptors on the T-cell surface. Subsequently, T-cell surface expression of an inhibitory molecule, CTLA-4, takes place. CTLA-4 competitively inhibits the binding of B7 to CD28 by interacting with the same ligands and prevents the co-stimulatory signal, dampening T-cell activation and proliferation. CTLA-4 thereby serves as a physiologic “brake” on the activated immune system [8-11].
Anti-PD-1 antibodies
A second co-inhibitory pathway uses the PD-1 receptor, which is another inhibitory receptor present on activated T cells. PD-1 is a protein that is encoded in humans by PDCD gene. PD-1 is a cell surface immune checkpoint receptor which belongs to the immunoglobulin super family and is expressed on T cells and pro-B cells. PD-1 binds to its two ligands, PD-L1 and PD-L2 which are the members of B7 family. PD-1 and its ligands play an important role in down regulating the immune system by preventing the activation of T cells, which in turn reduces autoimmunity and promotes self-tolerance. The inhibitory effect of PD-1 is accomplished through a dual mechanism of promoting apoptosis (programmed cell death) in antigen specific T cells in lymph nodes while simultaneously reducing apoptosis in regulatory T cells (suppressor T cells).
PD-1 binds to its ligands PD1-L1 (B7-H1) and PD1-L2 (B7-DC), which are expressed on tumor cells, thereby causing immunosuppression and preventing the immune system from rejecting the tumor. When PD-1 binds to its ligand (PD-L1) (often present on tumor cells), the ability of the activated T cell to produce an effective immune response is down-modulated. Monoclonal antibodies targeting both PD-1 and PD-L1 are being developed to interrupt this pathway and to augment the antitumor immune response; these have demonstrated significant clinical activity against several tumor types [12, 13].
| Ipilimumab | ▴Top |
Ipilimumab, an anti-CTLA-4 monoclonal antibody, is the first agent ever proven to improve survival in advanced melanoma. It was approved by Food and Drug Administration (FDA) in 2011. The current FDA approved dosing schedule for ipilimumab is 3 mg/kg intravenous (IV) infusion every 3 weeks (Q3W) for a total of four doses.
Efficacy
Ipilimumab prolongs the survival rate in patient with MM and this has been confirmed in two large randomized phase III trials.
The first study by Hodi et al included 676 patients with unresectable stage III or IV melanoma whose disease had progressed after previous treatment. The patients were randomized in a 3:1:1 ratio to receive ipilimumab with the melanoma peptide vaccine gp100 (n = 403), ipilimumab with gp100 placebo (n = 137), or gp100 vaccine with ipilimumab placebo (n = 136). Ipilimumab was dosed at 3 mg/kg IV Q3W for a total of four treatments with the vaccine administered immediately after each ipilimumab infusion. The median OS at 20 months was 10, 10.1, or 6.4 months for patients treated with the combination, ipilimumab alone, or gp100 alone, respectively. The increased survival in both ipilimumab containing regimens was statistically significant. One- and 2-year OS rates for ipilimumab alone were 45.6% and 23.5%, respectively; for gp100 alone, 25.3% and 13.7%, respectively; and for ipilimumab plus gp100, 43.6% and 21.6%, respectively [14].
In a second phase III study which was conducted by Robert et al, 502 treatment-naive patients with unresectable stage III or IV melanoma were randomly assigned to in 1:1 ratio to receive either dacarbazine with ipilimumab (n = 250) or dacarbazine with placebo (n = 252) given at weeks 1, 4, 7 , 10. Ipilimumab was administered at a higher dose of 10 mg/kg for four doses followed by a maintenance phase. The addition of ipilimumab to dacarbazine significantly improved the primary outcome of OS compared with dacarbazine alone (11.2 vs. 9.1 months; P < 0.05) [15].
A recent study published on February 9, 2015 was a pooled analysis of long-term survival data from phase II and III trials of ipilimumab in unresectable MM. The main purpose of the study was to give a precise estimate of long-term survival for ipilimumab-treated patients. The study included pooled OS data for 1,861 patients from 10 prospective and two retrospective studies of ipilimumab, including two phase III trials. Among 1,861 patients, median OS was 11.4 months (95% CI, 10.7 - 12.1 months), which included 254 patients with at least 3 years of survival follow-up. The survival curve began to plateau around year 3, with follow-up of up to 10 years. Three-year survival rates were 22%, 26%, and 20% for all patients, treatment-naive patients, and previously treated patients, respectively. These data once again support the durability of long-term survival in ipilimumab-treated patients with advanced melanoma [16].
Usage in other cancers
Ipilimumab is under various phases of clinical trials for the treatment of metastatic renal cell [17], prostate [18] and non-small cell lung cancers [19].
| Pembrolizumab | ▴Top |
Pembrolizumab is an anti-PD-1 monoloclonal antibody that has been extensively evaluated in ipilimumab-naive and previously treated melanoma patients. Pembrolizumab at a dose of 2 mg/kg IV Q3W was approved by the US FDA in September 2014 for patients who have progressed on treatment with ipilimumab.
Efficacy
Pembrolizumab was awarded FDA approval based on the following data from an international multicenter, open-label, randomized, dose-comparative phase 1 study randomizing 655 patients (342 ipilimumab treated (IPI-T) and 313 ipilimumab naive (IPI-N)) with unresectable or MM to receive pembrolizumab 2 mg/kg or 10 mg/kg IV once Q3W or 10 mg/kg every 2 weeks (Q2W). In the published analysis of 173 patients from this trial with ipilimumab-resistant disease who were assigned to either 2 mg/kg or 10 mg/kg Q3W, objective response rate (ORR) was achieved in 26% in both treatment arms with response duration lasting from 1.4 to 8.5 months (KEYNOTE-001 trial) [20].
The results of all the patients enrolled in this trial were recently published at American Society of Clinical Oncology (ASCO) 2015 annual meeting. ORR was 34% (29% IPI-T, 38% IPI-N), with a 6% complete remission (CR) rate. Median time to response was 2.8 months (range, 1.6 - 19.3). Eighty percent of responses were ongoing at the time of analysis, and median duration of response (DOR) was not reached (range, 6+ to 98+ weeks). Median progression-free survival (PFS) was 5.2 months (IPI-T, 4.9 months (3.0 - 5.5); IPI-N, 5.4 months (3.1 - 6.9)). PFS rates at 6 and 12 months were 44% and 34% (41% and 32% IPI-T, 47% and 36% IPI-N). The 1-year OS rate was 67%. Pembrolizumab provided durable antitumor activity, promising long-term survival data, and a manageable safety profile in both ipilimumab-naive and treated patients with MM [21].
In the KEYNOTE-002 trial which was a phase II trial, 540 patients with ipilimumab refractory MM were randomly assigned to pembrolizumab (2 mg/kg Q3W), pembrolizumab (10 mg/kg Q3W) or chemotherapy (carboplatin plus paclitaxel, paclitaxel alone, dacarbazine, or temozolomide per institutional standard) [22]. Treatment continued on this schedule until progressive disease. The ORRs (complete plus partial) were 21%, 26%, and 4% respectively, for pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg, and chemotherapy. Treatment was relatively well tolerated, with grade 3-5 adverse events (AEs) reported in 11% and 14% of the pembrolizumab treatment arms, and 26% of those managed with chemotherapy.
The ability of ipilimumab-refractory disease to respond to pembrolizumab is probably a reflection of the different mechanisms by which anti-CTLA-4 and anti-PD-1 therapies stimulate an anti-tumor T-cell response. CTLA-4 blockade broadens the immune response, evidenced by an increased T-cell receptor repertoire leading to increased tumor infiltration, whereas PD-1 blockade induces intratumoral T-cell proliferation without detectable changes in the peripheral immune repertoire.
Robert et al (KEYNOTE-006) have recently finished a randomized phase III trial of pembrolizumab in patients with advanced melanoma. Eight hundred thirty-four patients were assigned in a 1:1:1 ratio to receive pembrolizumab (at a dose of 10 mg/kg) Q2W or Q3W or four doses of ipilimumab (at 3 mg/kg) Q3W. The 6-month PFS rates were 47.3% for pembrolizumab Q2W, 46.4% for pembrolizumab Q3W, and 26.5% for ipilimumab. Estimated 12-month survival rates were 74.1%, 68.4%, and 58.2%, respectively. Treatment experienced adverse events (TEAEs) of grade 3-5 severity were lower in the pembrolizumab groups (13.3% and 10.1%) than in the ipilimumab group (19.9%) [23].
Usage in other cancers
Pembrolizumab has demonstrated highly durable response rates with minimal toxicity in large phase I studies involving patients with non-small-cell lung cancer (NSCLC), renal cell carcinoma, and other solid tumors [20, 24]. It has been FDA approved recently for the treatment of advanced NSCLC as a second-line treatment [25].
| Nivolumab | ▴Top |
Nivolumab is a monoclonal antibody that targets the PD-1 protein. Nivolumab at a dose of 3 mg/kg IV infused over 60 min Q2W was approved by US FDA in December 2014 for patients who progressed after treatment with ipilimumab and in patients whose tumors express BRAF V600 mutation.
Efficacy
In a phase I trial designed by Topalian et al, 107 patients were treated with nivolumab at doses from 0.1 to 10 mg/kg Q2W for up to 96 weeks. The results of this study showed a median survival of 17 months and CR or partial remission (PR) rates were observed in 34 of 107 patients (32%). One-year and 2-year survival rates were 62% and 43% respectively [26]. Expression of PD-L1 by the tumor appeared to predict for a higher response rate, long-term PFS, and long-term OS compared with tumors that did not express PD-L1. These results led to the conduction of further clinical trials.
In previously untreated patients, Robert et al conducted a phase III trial (Checkmate 066) (NCT01721772) [27]. Four hundred eighteen previously untreated MM patients without BRAF mutation were randomly assigned to nivolumab (at a dose of 3 mg/kg Q2W and dacarbazine-matched placebo Q3W) or dacarbazine (at a dose of 1000 mg/m2 of body-surface area Q3W and nivolumab-matched placebo Q2W). OS was significantly increased in those treated with nivolumab (1-year survival rate 73% versus 42%). PFS was also increased with nivolumab (median 5.1 versus 2.2 months), as was the ORR (40% versus 14%). Common AEs associated with nivolumab included fatigue, pruritus, and nausea. TEAEs of grade 3 or 4 occurred in 11.7% of the patients treated with nivolumab and 17.6% of those treated with dacarbazine.
In previously treated patients, Weber et al performed a randomized, controlled, open-label phase III trial to assess the efficacy and safety of nivolumab compared with chemotherapy as a second-line or later-line treatment in patients with MM (Checkpoint 037) (NCT01721746). All patients had received prior anti-CTLA-4 therapy and a BRAF inhibitor if a V600 mutation was present in their tumor. Two hundred seventy-two patients were randomly assigned to nivolumab group (3 mg/kg IV infusion Q2W) and 133 patients were assigned to chemotherapy group (either dacarbazine or carboplatin plus paclitaxel). The trial accrued 405 patients; preliminary results based upon 167 patients (120 treated with nivolumab and 47 with chemotherapy) showed that confirmed objective responses were significantly more common in patients treated with nivolumab compared with chemotherapy (32% versus 10%) [28]. Nivolumab led to a greater proportion of patients achieving an ORR and fewer toxic effects than with alternative available chemotherapy regimens for patients with advanced melanoma that has progressed after ipilimumab or ipilimumab and a BRAF inhibitor.
Usage in other cancers
Nivolumab has been studied to show some clinical activity in the treatment of Hodgkin’s lymphoma [29, 30], in patients with advanced, refractory NSCLC [31] and in patients with metastatic renal cell cancer [32]. Nivolumab is also being investigated under phase I/II study as a monotherapy or in combination with ipilimumab in locally advanced or metastatic tumors including triple negative breast, small cell lung, gastric and pancreatic malignancies (NCT01928394). Similarly, a phase IIb randomized study is evaluating the efficacy and safety of nivolumab alone or with ipilimumab when compared to bevacizumab in patients with recurrent glioblastoma (NCT02017717) [33]. Nivolumab has been FDA approved recently for the treatment of both squamous and non-squamous advanced, refractory NSCLC as a second-line treatment [34, 35].
| AEs of Immunotherapy | ▴Top |
Although the evolution of immune therapy antibodies can be associated with substantial benefits, by increasing immune system function, immune-checkpoint blockade can lead to inflammatory side effects called immune-related adverse events (IrAEs). IrAEs can affect any organ system, but they typically involve the skin, gastrointestinal, hepatic, and endocrine systems [36].
The most common IrAEs for both CTLA-4 and PD-1 therapy involve dermatologic toxicity. Physical examination findings can consist of a reticular, maculopapular, erythematous rash on the extremities or trunk [37]. Perhaps more unique to the PD-1 experience, oral mucositis and/or complaints of dry mouth have been reported in a small percentage of patients [26]. Fatigue is the most common side effect in patients with anti-PD-1 therapy.
Endocrinopathies affect the pituitary, thyroid, adrenal glands and manifest with non-specific symptoms such as nausea, headache, fatigue and vision changes. Diagnosis is usually made by characteristic laboratory findings and/or radiographic changes, such as enlargement of the pituitary gland [38].
Table 1 depicts the most common AEs and their grades.
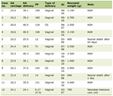 Click to view | Table 1. Adverse Effects of Immunotherapy and Management |
| Management of AEs | ▴Top |
For patients with moderate (grade 2) immune-mediated toxicities, treatment should be withheld and should not be resumed until symptoms resolve. Corticosteroids (prednisone 0.5 mg/kg/day or equivalent) should be started if symptoms do not resolve in a week. For patients with severe or life-threatening AEs, treatment should be stopped permanently and high-dose corticosteroids (prednisone 1 - 2 mg/kg/day or equivalent) should be given.
Patients who benefit from steroids generally do so within a few days. If symptoms do not improve after 3 days of treatment with IV steroids, next step is to administer infliximab (5 mg/kg) rather than continuing prolonged course of high-dose IV corticosteroids. In cases of severe hepatotoxicity, mycophenolate mofetil (500 mg orally every 12 h) can be administered concurrently with steroids and infliximab is contraindicated in such patients [39].
IrAEs associated with CTLA-4 blockade increase with increasing dose, whereas IrAEs associated with PD-1 blockade do not appear to be dose-related. Some IrAEs are also associated with the targeting of one pathway but not the other [26].
| Mechanism of Action of Targeted Therapy | ▴Top |
BRAF and MEK inhibitors
BRAF is a human gene that makes a protein called B-Raf. The B-Raf protein is involved in sending signals inside cells, which are involved in directing cell growth. In 2002, it was shown to be faulty (mutated) in some human cancers. B-Raf is a member of the Raf kinase family of growth signal transduction protein kinases. This protein plays a role in regulating the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (ERKs) signaling pathway, which affects cell division, differentiation, and secretion.
Mutations in BRAF gene can occur in two ways. It can be either inherited and cause birth defects or it can be acquired later in life and causes cancer. The frequency of BRAF mutations varies widely in human cancers, from more than 80% in melanomas and nevi, to as little as 0-18% in other tumors, such as 1-3% in lung cancers and 5% in colorectal cancer. In 90% of cases, thymine is substituted with adenine at nucleotide 1799 which leads to valine (V) being substituted for by glutamate (E) at codon 600 referred to as V600E mutation that activates the MAPK pathway. The V600K mutation results in an amino acid substitution at codon 600 in BRAF, from a valine (V) to lysine (K). This mutation has been widely associated with papillary thyroid cancer, NSCLC, colorectal cancer and melanoma.
The MAPK pathway plays an important role in the pathogenesis of melanoma. This pathway is physiologically activated when extracellular signals bind to their cognate membrane receptor, typically a receptor tyrosine kinase (RTK). Subsequently activated tyrosine receptor kinase leads to stimulation of small cytoplasmic proteins of RAS gene family (HRAS, NRAS and KRAS). Among them, NRAS mutations are found in about 10-15% of melanoma patients. Activated RAS results in a cascade of phosphorylation events involving the serine/threonine kinases RAF (encoded by ARAF, BRAF and CRAF). Activated RAF kinases phosphorylate and activate MEK1/2, which in turn phosphorylate and activate ERK1/2, leading to cellular proliferation, survival, and differentiation, and to an inhibitory feedback toward upstream components of the pathway (Fig. 1). About 50% of melanomas harbor an activating mutation in BRAF, the most common being BRAF V600E, which renders the kinase constitutively active [40-42].
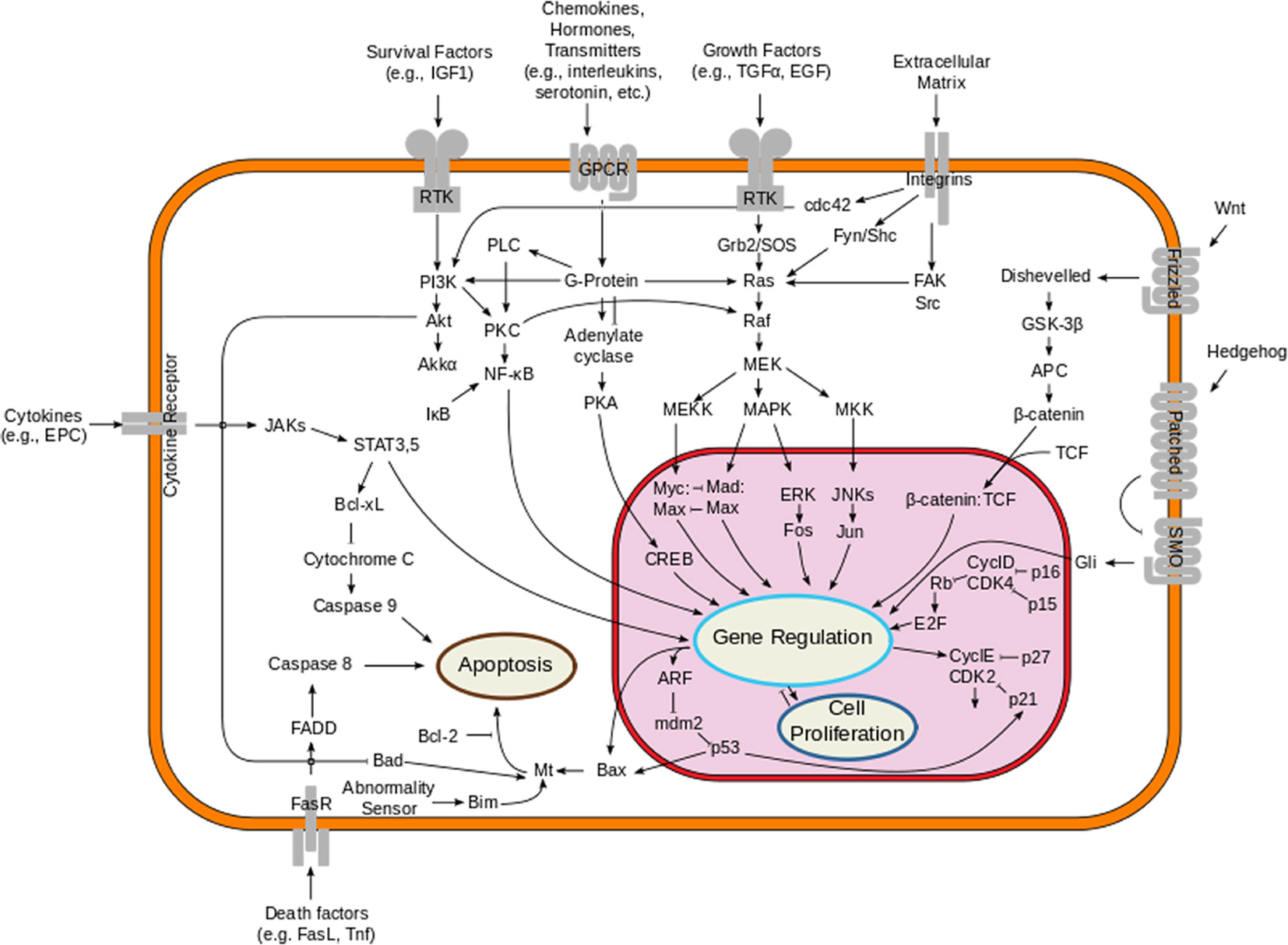 Click for large image | Figure 1. Overview of the signal transduction pathway like mitogen-activated protein kinase/extracellular signal-regulated kinases signaling pathway. |
| Vemurafenib | ▴Top |
Vemurafenib is a potent inhibitor of the kinase domain in mutant BRAF, a mutation carried by half of melanomas. It was approved by US FDA and European Medicines Agency for the treatment of unresectable or MM with mutant BRAF V600E at a dose of 960 mg orally twice a day.
Efficacy
A multicenter, phase I dose-escalation trial of 55 patients (49 with melanoma) established a phase II recommended dose of 960 mg twice daily [43]. In an extension phase of this trial, 32 patients with previously treated BRAF V600E-mutant MM demonstrated a tumor response rate of 81% (n = 26), with two patients demonstrating a CR. Among patients with symptomatic disease, improvement in symptoms was reported within 1 - 2 weeks.
A follow-up, multicenter, phase II study enrolled 132 patients with previously treated BRAF V600-mutant MM without brain metastases [44]. Patients who received vemurafenib 960 mg twice daily demonstrated an ORR of 53% (95% CI: 44-62%), with an additional 29% of patients achieving some degree of tumor control. The median PFS was 6.8 months and the median OS was 15.9 months.
In the international, multicenter, randomized, phase III BRIM-3 trial, 675 patients with previously untreated, MM with the BRAF V600E mutation were randomly assigned to either vemurafenib (960 mg twice a day) or dacarbazine (1,000 mg/m2 IV Q3W) in patients who had either metastatic disease or unresectable stage IIIC disease. The results of this study showed that OS was significantly prolonged with vemurafenib compared with dacarbazine (13.6 versus 9.7 months). PFS was also significantly prolonged (6.9 versus 1.6 months) [45]. Vemurafenib produced improved rates of OS and PFS in patients with previously untreated melanoma with the BRAF V600E mutation.
Development of resistance
Several mechanisms have been hypothesized regarding the development of resistance to vemurafenib after a certain period of treatment. Mechanisms of primary resistance include RAC1P29S mutations (RAC 1 regulates cell proliferation and migration), COT overexpression (COT activates ERK through mechanism that does not depend on RAF signaling), alterations in RTK signaling (RTK activation can signal either through CRAF or through the PI3K pathway), and alterations in the PI3K-AKT-mTOR pathway (loss of function of PTEN). The reactivation of the MAPK pathway is the most frequent cause of acquired/secondary resistance; it may be driven by events that occur upstream (upregulation and activation of the RTK, NRAS activating mutations) or downstream (activating MEK1/2 mutation, or at the level of BRAF) [46].
Another mechanism that has been proposed is the overexpression of eIF4E which is a translation initiation factor. eIF4E is overexpressed in a panel of melanoma cell lines, compared to immortalized melanocytes. Knock-down of eIF4E significantly repressed the proliferation of a subset of melanoma cell lines. Moreover, in BRAF V600E melanoma cell lines, vemurafenib inhibits 4E-BP1 phosphorylation, thus promoting its binding to eIF4E. Cap-binding and polysome profiling analysis confirmed that vemurafenib stabilizes the eIF4E-4E-BP1 association and blocks mRNA translation, respectively. Conversely, in cells with acquired resistance to vemurafenib, there is an increased dependence on eIF4E for survival, 4E-BP1 is highly phosphorylated and thus eIF4E - 4E-BP1 associations are impeded. Moreover, increasing eIF4E activity by silencing of 4E-BP1/2 renders vemurafenib responsive cells more resistant to BRAF inhibition [47].
AEs
The most common TEAEs were arthralgia, rash, nausea, photosensitivity, fatigue, cutaneous squamous cell carcinoma, pruritus and palmar-plantar dysesthesia in the phase I trial [43]. Increased incidence of squamous cell carcinoma in these patients resulted from increased proliferation of HRAS-mutant cell lines exposed to vemurafenib which was associated with a paradoxical re-activation of MAPK signaling [48]. A vemurafenib analog (PLX4720) accelerated the growth of the lesions harboring HRAS mutations, and this growth was inhibited by concomitant treatment with an MEK inhibitor. The common AEs in phase II and phase III trials were cutaneous events, arthralgia, rash, fatigue, alopecia, photosensitivity, nausea and diarrhea [44, 45].
A few case reports have been published in the literature where vemurafenib is associated with development of uveitic cystoid macular edema [49], Fanconi syndrome [50], gingival hyperplasia [51], peripheral facial palsy [52], severe radiation dermatitis [53] and prolongation of QT interval.
| Dabrafenib | ▴Top |
Dabrafenib is another BRAF kinase inhibitor that has demonstrated significant activity in patients with advanced melanoma compared with dacarbazine chemotherapy. Dabrafenib was approved by the US FDA in May 2013 for the treatment of patients with advanced melanoma that contains the V600E mutation of BRAF at a dose of 150 mg orally twice a day.
Efficacy
An initial phase I trial of patients with BRAF V600E-mutant melanoma examined escalating doses of dabrafenib in 184 patients (156 patients with MM and 28 with non-melanoma solid tumors) [54, 55]. This trial established a dose of 150 mg orally twice daily for further studies.
A phase II study (BREAK-2) evaluated the use of dabrafenib in patients with BRAF-mutant treatment-naive MM. A total of 76 patients with BRAF V600E and 16 patients with V600K mutations were treated with dabrafenib 150 mg twice daily. BRAF V600E patients had a confirmed response rate of 59% (95% CI: 48-70%) with a 7% CR rate (n = 5), whereas patients with the BRAF V600K mutation had a response rate of 13% (95% CI: 0-28%). Median OS was 13.1 months for the V600E patients and 12.9 months for the V600K patients [56].
An open-label, multicenter phase II trial was conducted in patients with brain metastases to assess the efficacy of dabrafenib (BREAK-MB) [57]. One hundred seventy-two patients were enrolled and split into two cohorts; those with no prior local treatment for brain metastases (cohort A, n = 89) and those who had progressed after previous local treatment (cohort B, n = 83). Patients were treated with 150 mg of dabrafenib twice daily. An intracranial response was observed in 39% (95% CI: 28-51%) in cohort A and 30% (95% CI: 20-43%) in cohort B patients.
In the pivotal phase III trial, 250 previously untreated patients with unresectable stage III or stage IV melanoma were randomly assigned in a 3:1 ratio to either dabrafenib (150 mg orally twice a day) or dacarbazine (1,000 mg/m2 IV Q3W). All patients had the V600E mutation in BRAF, the patients in the dacarbazine arm being allowed to crossover at the time of disease progression. Dabrafenib significantly improved median PFS to 5.1 months as compared to 2.7 months for dacarbazine (hazard ratio (HR) = 0.30, P < 0.001). The confirmed response rate was 50% for dabrafenib, with a 3% (n = 6) CR rate, and a median time to response of 6.3 weeks [58]. Dabrafenib significantly improved PFS compared with dacarbazine.
AEs
The most common grade 2 or higher AEs in the phase I trial were cutaneous squamous cell carcinoma or keratoncanthoma, fatigue and pyrexia [54]. Common toxicities in the phase II trial (BREAK-2) included arthralgia, hyperkeratosis, pyrexia, fatigue and headache. Serious AEs were reported in 25 patients (27%) and these included basal cell carcinoma, cutaneous squamous cell carcinoma, anemia, pyrexia, non-cardiac chest pain and vomiting [56]. The common AEs in phase III trial were cutaneous events, rash, fatigue, headache and arthralgia [58]. Dose reduction was required in 28% (n = 52) of patients, and five patients (3%) discontinued the drug.
Usage in other cancers
Dabrafenib is also used in the treatment of hairy cell leukemia along with melanoma [59]. Dabrafenib is used in the treatment of melanoma in patients with leucopenia induced by vemurafenib as dabrafenib has no negative influence on leucocyte count [60]. Dabrafenib can be used as a substitute for vemurafenib if the patient develops severe cutaneous AE like toxic epidermal necrolysis from vemurafenib requiring its discontinuation [61]. Dabrafenib was well tolerated and resulted in durable responses in BRAF-mutant differentiated thyroid carcinoma patients [62].
| Trametinib | ▴Top |
Trametinib, an MEK inhibitor, was first approved by FDA in 2013 for treatment of patients with unresectable or MM with BRAF V600E or V600K mutation. Trametinib is usually taken as 2 mg orally once daily as a single agent or 2 mg taken orally once daily with dabrafenib 150 mg orally taken twice.
Efficacy
Based on the below mentioned trial, trametinib was approved by the FDA in 2013 for the treatment of patients with unresectable stage IIIC or MM with BRAF V600E/K mutations.
In an open-label phase III trial (METRIC), 322 patients with BRAF V600E/K-mutant MM were randomized to receive either trametinib (n = 214) or chemotherapy with either dacarbazine or paclitaxel (n = 108) [63]. Patients who received prior BRAF inhibitors or MEK inhibitors were excluded from study. Patients in the chemotherapy group were allowed to crossover to trametinib with disease progression. The primary endpoint is median PFS which was improved to 4.8 months in the trametinib group, as compared to 1.4 months in the chemotherapy group (HR = 0.45, 95% CI: 0.33 - 0.63; P < 0.001). The OS rate at 6 months was 81% in the trametinib group and 67% in the chemotherapy group (HR = 0.54, 95% CI: 0.32 - 0.92; P = 0.01), despite 65% of patients in the chemotherapy arm crossing over to the trametinib arm.
Prior to this trial, there was a phase I trial looking at pharmacodynamics, dose escalation and AEs.
The phase I trial study was done in three parts: firstly, dose escalation to define the maximum tolerated dose (MTD) followed by identification of the recommended phase II dose, and lastly assessment of pharmacodynamic changes. The study included 206 patients who had any solid tumors, of which 97 patients were diagnosed with MM. Blood samples and tumor biopsy specimens were taken to assess pharmacokinetic and pharmacodynamic changes. AEs were defined with common toxicity criteria, and tumor response was measured by response evaluation criteria in solid tumors (RECIST). Overall ORR was 10%. However, BRAF-mutant melanoma had a response rate of 33% [64]. The RECIST-defined response rate was 40% in the 30 patients with BRAF inhibitor-naive MM, but only 17% in those with prior BRAF inhibitor therapy (n = 6). This study has recommended a dose of 2 mg once a day with manageable side effects.
AEs and safety
The most common AEs with trametinib are skin-related toxicities followed by diarrhea and less common included ocular toxicities and decrease in left ventricular ejection fraction. These are explained in detail below.
In the phase I dose-escalation study of trametinib, a loading dose regimen of two 10 mg/day loading doses followed by 3 mg/day led to grade 3 diarrhea, grade 3 rash and grade 2 central serous retinopathy. Of the 70 patients treated with the phase II recommended dose of 2 mg/day, only eight (11%) had grade 3 treatment-related events requiring dose reduction. The most typical event leading to dose reduction was rash, which was poorly classified but most often was acneiform [54, 64]. Although skin-related toxicities were common, no events of cutaneous squamous cell carcinoma or other proliferative skin lesions were recorded. Diarrhea was predominately grade 1 and was manageable with standard symptom-based therapies. Treatment-related ocular toxicities were recorded in 31 patients (15%), including one episode of retinal vein occlusion at the phase II recommended dose of 2 mg/day. The visual acuity of this patient improved after intraocular treatments of antibodies against VEGF. A decline in left ventricular ejection fraction was noted in 16 (8%) patients, but most of these events were grade 2 or lower. The mechanism of reduced left ventricular ejection fraction in relation to MEK inhibition is unknown, but cardiac toxicity has previously been reported in association with MEK inhibitors; this could represent a class effect [65]. Subject’s cardiac function did return to baseline after discontinuation of trametinib.
In the phase III trial, the common AEs associated with trametinib therapy were rash, diarrhea, peripheral edema, fatigue and dermatitis [63]. A decrease in left ventricular ejection fraction was noted in 14 patients (7%) and two patients (1%) had serious cardiac-related events that were considered to be drug-related, prompting drug discontinuation. Ocular events (mostly grade 1 or 2) occurred in 9% of patients, with no observed cases of retinal vein occlusion. In total, AEs led to treatment interruption in 35% of patients and to dose reduction in 27%.
| Combination Therapy | ▴Top |
MAPK pathway inhibition by either BRAF or MEK inhibitors has proven to increase PFS and OS in patients with MM. Their efficacy has been curtailed by the toxicity of the side effects and development of resistance in patients as well as the emergence of secondary cancers such as cutaneous squamous cell carcinoma due to the activation of BRAF inhibitor-induced paradoxical activation of MAPK pathway. Combination therapy with BRAF and MEK inhibitors is displaying potential to overcome these roadblocks. The studies below demonstrate that the combination therapies decrease the advent of secondary cancers, while delaying the emergence of resistance and increasing the PFS and OS compared to monotherapy [66, 67].
| Combined BRAF and MEK Inhibitor | ▴Top |
Dabrafenib and trametinib versus dabrafenib and placebo
In a phase III double-blinded study conducted by Long et al [68], 423 patients with unresectable stage IIIC or stage IV melanoma with BRAF V600E/K mutation were randomized to receive the combination of oral BRAF inhibitor dabrafenib and MEK inhibitor trametinib (n = 211) (combination therapy group) or dabrafenib and placebo (n = 212) (dabrafenib group). Patients who had previous systemic anticancer treatment and those without the BRAF V600E/K mutation were excluded from the study. The patients were stratified according to the BRAF genotype and baseline LDH level. The primary endpoint was PFS and the secondary endpoints were OS, response rate, response duration, safety and pharmacokinetics.
Efficacy
In the intention-to-treat group, the estimated median PFS was longer in the combination therapy group compared to the dabrafenib only group (9.3 vs. 8.8 months with HR of 0.75, P = 0.03). In the patient population with elevated LDH levels, the median PFS was longer in the combination therapy group compared to monotherapy (7.1 vs. 3.8 months). The ORR was 67% in the combination therapy group versus 51% in the monotherapy group. Ten percent of patients achieved CR and 56% had PR in the dabrafenib-trametinib group compared to 9% of patients with CR and 43% with PR in the dabrafenib only group.
Quality of life and pain management are important factors in patients with non-curative cancers such as MM. The study done by Schadendorf et al showed that the combination therapy is shown to have superior conservation of health-related quality of life and improvement in pain compared to monotherapy [69].
AEs
In both combination therapy and monotherapy groups, pyrexia, fatigue, headache, nausea, diarrhea and arthralgia were the most commonly reported AEs. However, pyrexia, hypertension, peripheral edema and diarrhea were more common in the combination group than the monotherapy group. On the other hand, the incidence of alopecia, papillomas and hand and foot syndrome was lower in the dabrafenib-trametinib group versus dabrafenib alone. Secondary cancers such as cutaneous squamous cell carcinoma were also lower in the combination therapy group vs. monotherapy (2% vs. 9%).
Vemurafenib and cobimetinib versus vemurafenib and placebo
A phase III randomized trial conducted by Larkin et al [70] on 495 patients with BRAF V600E/K-mutated metastatic or locally invasive melanoma showed similar results to the above study. Patients who had received prior cancer treatment and those with wild type BRAF mutation were excluded from the study. Patients were randomly assigned to receive either combination therapy of oral BRAF inhibitor vemurafenib (960 mg twice daily) along with oral MEK inhibitor cobimetinib (60 mg once daily) or monotherapy of oral vemurafenib along with placebo. The primary endpoint was PFS and the secondary endpoints were OS, response rate, response duration and safety. The results reported are from July 2014.
Efficacy
The combination group had a significantly higher PFS compared to the control group (9.9 vs. 6.2 months, 95% CI: 5.6 - 7.4). The HR was determined to be 0.51 (95% CI: 0.39 - 0.68; P < 0.001). The OS at 9 months for the combination group was 81% compared with 73% of the control group. The response rate was determined to be higher in the combination group compared to the control group 68% vs. 45% (P < 0.001). CR was also higher in the combination group vs. control group (10% vs. 4%) and the median response duration was 7.3 months in the combination group while the control group did not reach the median.
AEs
Central serous retinopathy, elevated aminotransferase, creatinine kinase levels and gastrointestinal symptoms occurred at a higher rate in the combination group compared to the control group. The majority of these events were either grade 1 or 2.
Ipilimumab plus sargramostim vs. ipilimumab
Ipilimumab is an IgG1 monoclonal antibody that blocks CLTA-4 thereby enhancing T-lymphocyte activity. Sargramostim is a granulocyte monocyte colonoy stimulating factor (GM-CSF) cytokine that augments antigen presentation by dendritic cells thereby improving lymphocyte antitumor activity [11]. In a phase II randomized clinical trial study by Hodi et al [71], 245 patients with stage III or IV melanoma with at least one prior therapy were randomized to receive CTLA-4 blockade with ipilimumab and GM-CSF secreting tumor vaccine sargramostim (n = 123) or ipilimumab (n = 122) alone. The primary objective was OS and the secondary endpoints were PFS, response rate, safety and the tolerability. Median time of follow-up was 13.3 months. The OS data as of December 2012 and other data as of March 2013 were reported.
Efficacy
The median OS for the combination group was 17.5 months (95% CI: 14.9-not reached) versus 12.7 months (95% CI: 10.0-not reached) for the monotherapy group. The 1-year survival rate for the ipilimumab-sargramostim group was 68.9% (95% CI: 60.6-85.5%) compared to 52.9% (95% CI: 43.6-62.2%) for the ipilimumab only group. Subgroup analysis showed that men who were treated with ipilimumab plus sargramostim had greater OS compared to ipilimumab alone while the opposite trend was seen in women. Caution should be used when interpreting the subgroup results as the sample size and number of deaths in subgroups was comparatively small. However in PFS and response rate, there was no statistical difference between the two groups.
AEs
Grade 3-5 events occurred in 44.9% of ipilimumab-sargramostim group compared to 58.3% in the ipilimumab group. Gastrointestinal toxicities such as colonic perforation and pulmonary toxicity were notably decreased in the ipilimumab-sargramostim group versus the ipilimumab group (16.1% vs. 26.7% and 0% vs.7.5%) respectively.
Other combinations
Vemurafenib and ipilimumab
In a phase I study conducted by Ribas et al [72], the first cohort of six patients each received full dose of vemurafenib (960 mg twice daily) for 1 month followed by ipilimumab infusions Q3W along with twice daily dose vemurafenib. Four of the six patients developed dose-limiting, grade 3 increase in aminotransferase levels 2 - 5 weeks after first infusion of ipilimumab.
A second cohort of six patients received decreased dose of vemurafenib (720 mg twice daily) with full dose of ipilimumab. Two of the patients developed grade 3 elevations and one developed grade 2 elevation of aminotransferase 3 weeks after ipilimumab infusion. The study was discontinued secondary to hepatotoxicity of the combination therapy.
Nivolumab plus ipilimumab
In a phase I trial conducted by Wolchok et al [73], 53 patients received anti-CTLA-4 antibody ipilimumab and anti-PD-1 receptor antibody nivolumab concurrently while 33 patients received sequenced treatment. All patients had stage III or IV unresectable melanoma. Ninety-three percent of patients in concurrent group had TEAEs compared to 73% in the sequenced treatment group. Most common AEs were rash, pruritus, fatigue and diarrhea. Dose-limiting grade 3 or 4 events were noted in 21% of patients in the concurrent group vs. 9% in the sequenced group. Most drug-related events were treated with immunosuppressant. The data from this study showed that the combination therapy had an acceptable safety profile and warrants further investigation to compare efficacy of the combination therapy versus monotherapy in advanced melanoma.
Postow et al [74] recently conducted a double-blinded study involving 142 patients (109 with BRAF wild type and 33 with BRAF V600E mutation positive) with MM who had not previously received treatment. They were randomly assigned in a 2:1 ratio to receive ipilimumab (3 mg/kg body weight) combined with either nivolumab (1 mg/kg) or placebo once Q3W for four doses, followed by nivolumab (3 mg/kg) or placebo Q2W until the occurrence of disease progression or unacceptable toxic effects. The primary endpoint was the rate of investigator-assessed, confirmed objective response among patients with BRAF V600 wild-type tumors.
Among patients with BRAF wild-type tumors, the rate of confirmed objective response was 61% (44 of 72 patients) in the group that received both ipilimumab and nivolumab (combination group) versus 11% (four of 37 patients) in the group that received ipilimumab and placebo (ipilimumab-monotherapy group) (P < 0.001), with CR reported in 16 patients (22%) in the combination group and no patients in the ipilimumab-monotherapy group. The median duration of response was not reached in either group. The median PFS was not reached with the combination therapy and was 4.4 months with ipilimumab monotherapy. Similar results for response rate and PFS were observed in 33 patients with BRAF mutation-positive tumors. Drug-related AEs of grade 3 or 4 were reported in 54% of the patients who received the combination therapy as compared with 24% of the patients who received ipilimumab monotherapy. The ORR and the PFS among patients with advanced melanoma who had not previously received treatment were significantly greater with nivolumab combined with ipilimumab than with ipilimumab monotherapy.
Recently Larkin et al [75] conducted a double-blinded, phase III trial comparing nivolumab alone or nivolumab plus ipilimumab to ipilimumab alone in untreated patients with advanced MM. Nine hundred forty-five patients were randomly assigned in a 1:1:1 ratio to either nivolumab alone or nivolumab plus ipilimumab or ipilimumab alone. The primary endpoints were PFS and OS. The median PFS was 11.5 months in patients with nivolumab plus ipilimumab, 2.9 months in ipilimumab alone (P < 0.001) and 6.9 months in nivolumab alone (P < 0.001). In patients with tumors positive for PD-L1, the median PFS was 14.0 months in both nivolumab plus ipilimumab and nivolumab alone groups, whereas in patients with PD-L1 negative tumors, PFS was longer with combination therapy than nivolumab alone. TEAEs of grade 3 or 4 occurred in 16.3% of the patients in the nivolumab group, 55.0% of those in the nivolumab-plus-ipilimumab group, and 27.3% of those in the ipilimumab group.
This study proved that in previously untreated patients with MM, nivolumab alone or nivolumab with ipilimumab resulted in significantly longer PFS than ipilimumab alone. In patients with PD-L1-negative tumors, the combination of PD-1 and CTLA-4 blockade was more effective than either agent alone.
| Conclusion and Approach to Therapy | ▴Top |
In summary, the survival rate of patients with MM far improved by the development of these novel drugs and drug combinations. Anti-PD-1 antibodies like pembrolizumab and nivolumab have become the preferred approach to immunotherapy in patients with advanced melanoma, even though they are associated with various autoimmune AEs. Anti-CTLA-4 antibody like ipilimumab retains a role in combination with anti-PD-1 antibodies. Targeted therapy against MAPK pathway is an important option for the treatment of patients with characteristic BRAF V600 mutation. Targeted therapy is not indicated in patients without a characteristic V600 mutation.
The choice to use either immunotherapy or targeted therapy or combination therapy depends on the performance status of the patient and BRAF V600 mutation.
For patient with a BRAF V600 mutation and good performance status, immunotherapy is recommended initially rather than targeted therapy. Immunotherapy in the form of nivolumab and ipilimumab combination is recommended as the initial systemic therapy. For those patients whose disease can no longer be controlled with this immunotherapy, targeted therapy using a combination of BRAF inhibitor/MEK inhibitor (dabrafenib/trametinib) is recommended.
For patients with BRAF V600 mutation and bulky disease, elevated serum LDH, visceral metastases, poor performance status, targeted therapy is initially recommended. Immunotherapy is also an alternative and may be recommended after progression on targeted therapy in those patients.
For patients without BRAF V600 mutation and good performance status, immunotherapy that includes combination of nivolumab and ipilimumab is initially recommended. Anti-PD-1 monotherapy with either nivolumab or pembrolizumab is a suitable alternative when toxicity is a concern with combination therapy. For poor performance status patients without BRAF V600 mutation, who are not thought to be able to tolerate combination treatment, single agent anti-PD-1 therapy is recommended.
The advent of immune and targeted therapy has ushered in an era of optimism for the treatment of MM. Monotherapy regimens with these new pharmaceuticals are hampered by the emergence of dose-limiting toxicity and secondary cancers. Although some of these concerns are addressed by the combination therapies, further studies are needed to evaluate triple combination therapies and new immune modulators and targeted therapy.
In summary, the treatment of advanced-staged melanoma is a rapidly developing field. Several new and effective treatments have been introduced in recent years, to the benefit of melanoma patients of all ages. With an increasing incidence of melanoma, particularly among older patients, their participation in clinical trials is essential.
| References | ▴Top |
- www.cancer.org. American Cancer Society, Cancer Fact and Figures. 2015.
- Macdonald JB, Dueck AC, Gray RJ, Wasif N, Swanson DL, Sekulic A, Pockaj BA. Malignant melanoma in the elderly: different regional disease and poorer prognosis. J Cancer. 2011;2:538-543.
doi pubmed - www.cancer.org. National Cancer Institute. Melanoma Treatment.
- Miller AJ, Mihm MC, Jr. Melanoma. N Engl J Med. 2006;355(1):51-65.
doi pubmed - Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT, et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5(7):1701-1752.
doi pubmed - de Souza CF, Morais AS, Jasiulionis MG. Biomarkers as key contributors in treating malignant melanoma metastases. Dermatol Res Pract. 2012;2012:156068.
doi - La-Beck NM, Jean GW, Huynh C, Alzghari SK, Lowe DB. Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacotherapy. 2015;35(10):963-976.
doi pubmed - Boasberg P, Hamid O, O'Day S. Ipilimumab: unleashing the power of the immune system through CTLA-4 blockade. Semin Oncol. 2010;37(5):440-449.
doi pubmed - Camacho LH. Novel therapies targeting the immune system: CTLA4 blockade with tremelimumab (CP-675,206), a fully human monoclonal antibody. Expert Opin Investig Drugs. 2008;17(3):371-385.
doi pubmed - Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7(2):95-106.
doi pubmed - Weber J. Immune checkpoint proteins: a new therapeutic paradigm for cancer--preclinical background: CTLA-4 and PD-1 blockade. Semin Oncol. 2010;37(5):430-439.
doi pubmed - Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
doi pubmed - Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
doi pubmed - Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
doi pubmed - Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517-2526.
doi pubmed - Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015;33(17):1889-1894.
doi pubmed - Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, Suri KB, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825-830.
doi pubmed - Fong L, Kwek SS, O'Brien S, Kavanagh B, McNeel DG, Weinberg V, Lin AM, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69(2):609-615.
doi pubmed - Tomasini P, Khobta N, Greillier L, Barlesi F. Ipilimumab: its potential in non-small cell lung cancer. Ther Adv Med Oncol. 2012;4(2):43-50.
doi pubmed - Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109-1117.
doi - Daud A RA, Robert C, Hodi S, Wolchok J, Joshua A et al. Long-term efficacy of pembrolizumab (MK-3475) in a pooled analysis of 655 patients with advanced melanoma enrolled in KEYNOTE-001. ASCO annual meeting2015.
- Ribas A PI, et al. A Randomized Controlled Comparison of Pembrolizumab and Chemotherapy in Patients With Ipilimumab-Refractory Melanoma. 2014 Society of Melanoma Rearch Conference.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521-2532.
doi pubmed - Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134-144.
doi pubmed - Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018-2028.
doi pubmed - Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020-1030.
doi pubmed - Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320-330.
doi pubmed - Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375-384.
doi - Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372(4):311-319.
doi pubmed - Bagcchi S. Nivolumab shows clinical activity in Hodgkin's lymphoma. Lancet Oncol. 2015;16(3):e108.
doi - Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16(3):257-265.
doi - Phillips R. Bladder cancer: Stem cells repopulate tumours. Nat Rev Urol. 2015;12(2):63.
doi pubmed - Gunturi A, McDermott DF. Nivolumab for the treatment of cancer. Expert Opin Investig Drugs. 2015;24(2):253-260.
doi pubmed - Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627-1639.
doi pubmed - Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(2):123-135.
doi pubmed - Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
doi pubmed - Lacouture ME, Wolchok JD, Yosipovitch G, Kahler KC, Busam KJ, Hauschild A. Ipilimumab in patients with cancer and the management of dermatologic adverse events. J Am Acad Dermatol. 2014;71(1):161-169.
doi pubmed - Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, Royal RE, et al. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother. 2005;28(6):593-598.
doi pubmed - Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm. 2009;24(3):321-325.
doi pubmed - Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401-1409.
doi pubmed - Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949-954.
doi pubmed - Solus JF, Kraft S. Ras, Raf, and MAP kinase in melanoma. Adv Anat Pathol. 2013;20(4):217-226.
doi pubmed - Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809-819.
doi pubmed - Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707-714.
doi pubmed - Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
doi pubmed - Spagnolo F, Ghiorzo P, Orgiano L, Pastorino L, Picasso V, Tornari E, Ottaviano V, et al. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015;8:157-168.
doi pubmed - Zhan Y, Dahabieh MS, Rajakumar A, Dobocan MC, M'Boutchou MN, Goncalves C, Shiru LL, et al. The role of eIF4E in response and acquired resistance to vemurafenib in melanoma. J Invest Dermatol. 2015;135(5):1368-1376.
doi pubmed - Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207-215.
doi pubmed - Fonollosa A, Mesquida M, Adan A. Uveitic macular oedema after treatment with vemurafenib. Acta Ophthalmol. 2015.
doi pubmed - Denis D, Franck N, Fichel F, Levi C, Dupin N. Fanconi syndrome induced by vemurafenib: a new renal adverse event. JAMA Dermatol. 2015;151(4):453-454.
doi pubmed - Mangold AR, Bryce A, Sekulic A. Vemurafenib-associated gingival hyperplasia in patient with metastatic melanoma. J Am Acad Dermatol. 2014;71(5):e205-206.
doi pubmed - Klein O, Ribas A, Chmielowski B, Walker G, Clements A, Long GV, Kefford RF. Facial palsy as a side effect of vemurafenib treatment in patients with metastatic melanoma. J Clin Oncol. 2013;31(12):e215-217.
doi pubmed - Satzger I, Degen A, Asper H, Kapp A, Hauschild A, Gutzmer R. Serious skin toxicity with the combination of BRAF inhibitors and radiotherapy. J Clin Oncol. 2013;31(13):e220-222.
doi pubmed - Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893-1901.
doi - Rheault TR, Stellwagen JC, Adjabeng GM, Hornberger KR, Petrov KG, Waterson AG, Dickerson SH, et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med Chem Lett. 2013;4(3):358-362.
doi pubmed - Ascierto PA, Minor D, Ribas A, Lebbe C, O'Hagan A, Arya N, Guckert M, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205-3211.
doi pubmed - Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, Puzanov I, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
doi - Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358-365.
doi - Blachly JS, Lozanski G, Lucas DM, Grever MR, Kendra K, Andritsos LA. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw. 2015;13(1):9-13; quiz 13.
pubmed - Orouji E, Ziegler B, Umansky V, Gebhardt C, Utikal J. Leukocyte count restoration under dabrafenib treatment in a melanoma patient with vemurafenib-induced leukopenia: case report. Medicine (Baltimore). 2014;93(28):e161.
doi pubmed - Jeudy G, Dalac-Rat S, Bonniaud B, Hervieu A, Petrella T, Collet E, Vabres P. Successful switch to dabrafenib after vemurafenib-induced toxic epidermal necrolysis. Br J Dermatol. 2015;172(5):1454-1455.
doi pubmed - Falchook GS, Millward M, Hong D, Naing A, Piha-Paul S, Waguespack SG, Cabanillas ME, et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid. 2015;25(1):71-77.
doi pubmed - Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107-114.
doi pubmed - Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773-781.
doi - Robert C, Dummer R, Gutzmer R, Lorigan P, Kim KB, Nyakas M, Arance A, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14(8):733-740.
doi - Aris M, Barrio MM. Combining immunotherapy with oncogene-targeted therapy: a new road for melanoma treatment. Front Immunol. 2015;6:46.
doi pubmed - Mallarkey G, Coombes RC. Targeted therapies in medical oncology: successes, failures and next steps. Ther Adv Med Oncol. 2013;5(1):5-16.
doi pubmed - Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877-1888.
doi pubmed - Schadendorf D, Amonkar MM, Stroyakovskiy D, Levchenko E, Gogas H, de Braud F, Grob JJ, et al. Health-related quality of life impact in a randomised phase III study of the combination of dabrafenib and trametinib versus dabrafenib monotherapy in patients with BRAF V600 metastatic melanoma. Eur J Cancer. 2015;51(7):833-840.
doi pubmed - Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867-1876.
doi pubmed - Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, Leming P, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744-1753.
doi pubmed - Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365-1366.
doi pubmed - Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122-133.
doi pubmed - Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006-2017.
doi pubmed - Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23-34.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.