

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://www.jocmr.org |
Review
Volume 15, Number 2, February 2023, pages 76-83

Current and Novel Therapeutical Approaches of Classical Homocystinuria in Childhood With Special Focus on Enzyme Replacement Therapy, Liver-Directed Therapy and Gene Therapy
Stefan Bittmanna, b, Gloria Villalona, Elena Moschuring-Alievaa, Elisabeth Luchtera, Lara Bittmanna
aPed Mind Institute, Department of Pediatrics, Medical and Finance Center Epe, D-48599 Gronau, Germany
bCorresponding Author: Stefan Bittmann, Ped Mind Institute, Department of Pediatrics, Medical and Finance Center Epe, D-48599 Gronau, Germany
Manuscript submitted November 12, 2022, accepted January 9, 2023, published online February 28, 2023
Short title: Therapeutical Approaches of Classical HCU
doi: https://doi.org/10.14740/jocmr4843
- Abstract
- Introduction
- Current and Novel Therapy Options
- Current Treatment Options of HCU
- Novel Therapeutic Approaches to Treat HCU
- Conclusions
- References
| Abstract | ▴Top |
Classical homocystinuria is a hereditary defect of the enzyme cystathionine beta synthase, which is produced in the liver. If this enzyme fails, the synthesis pathway of cysteine from methionine is interrupted, leading to the accumulation of homocysteine in the blood plasma and homocysteine in the urine. After birth, the children are unremarkable except for the characteristic laboratory findings. Symptoms rarely appear before the second year of life. The most common symptom is a prolapse of the crystalline lens. This finding is seen in 70% of untreated 10-year-old affected individuals. As the earliest symptom, psychomotor retardation occurs in the majority of patients already during the first two years of life. Limiting factors in terms of life expectancy are thromboembolism, peripheral arterial disease, myocardial infarction, and stroke. These symptoms are due to the damage to the vessels caused by the elevated amino acid levels. About 30% suffer a thromboembolic event by the age of 20, about half by the age of 30. This review focus on present and new therapeutical approaches like the role of enzyme replacement with presentation of different novel targets in research like pegtibatinase, pegtarviliase, CDX-6512, erymethionase, chaperones, proteasome inhibitors and probiotic treatment with SYNB 1353. Furthermore, we analyze the role of liver-directed therapy with three dimensional (3D) bioprinting, liver bioengineering of liver organoids in vitro and liver transplantation. The role of different gene therapy options to treat and cure this extremely rare disease in childhood will be discussed.
Keywords: Homocystinuria; Children; Novel treatment
| Introduction | ▴Top |
Classical homocystinuria (HCU) is a genetic disease based on a defect of cystathionine beta synthase as one important enzyme of transsulfuration pathway due to different underlying molecular mechanism, around 190 to date, which show different expression [1-58]. The mutation is found on the long arm of chromosome 21 (21q22.3) [1-58]. Deletions, mutations with frameshift aspect, codons with premature termination and different miss-splicing variants were described [1-51].
Overall, the disease is a defect in methionine metabolism [1-58]. Some mutations are pyridoxine responsive, some not [1-58]. Newborn screening shows often false negative results, so a new development, the paper spray ionization mass spectrometry (PSI-MS), was introduced to detect this rare disease in childhood [24, 35, 49, 50]. Per year, nearly four children with this rare disease will be born worldwide (birth rate 800,000/year), whereas an incidence of 1.1800 (Quatar) was described and differs in a wide range geographically [1-58]. Patients are homozygote or compound heterozygote [1, 4, 7, 16, 23, 42, 43]. Result of the disease is a disturbance of transsulfuration pathway with high homocysteine and methionine levels in blood [1-58]. Cystathionine beta deficiency presents with many different symptoms and pathological features, particularly affecting the eye, skeleton deformities, central nervous system (CNS) disturbances and vascular diseases. At birth, the children are still normal. If untreated, the disease is progressive. Symptoms in the eyes, an ocular hallmark, include ectopia lentis (50% of cases) with disruption of zonular fasers and high-grade myopia [1-51, 59, 60]. Skeletal symptoms include genu valgum, dolichostenomelia, moreover pectus excavatum or carinatum, and osteoporosis [1-58]. The most prominent cause of morbidity and mortality is thromboembolism of the great and small arteries and veins. Decreased intelligence rarely becomes manifest in the first or second year of life. Clinically significant psychiatric problems are present in 50% of patients. However, other organs, including the liver, hair, and skin, also show symptoms of the disease. Clinical diagnosis of cystathionine beta deficiency is confirmed by analysis of plasma amino acids (including total homocysteine), measurement of cystathionine beta synthase activity, or detection of cystathionine beta synthase mutations. If cystathionine beta synthase deficiency is diagnosed in the newborn, the desirable ideal case, then complications of the eyes and skeleton and thromboembolic events must be prevented, and the development of normal intelligence must be aimed for. If diagnosed late, the clinician’s goal must be to prevent life-threatening thromboembolic events and further deterioration of complications that have already occurred.
| Current and Novel Therapy Options | ▴Top |
There are different treatment modalities and ongoing research studies concentrating on new treatment options (Table 1) [21, 28, 37, 39, 47, 52-58, 61-79]. One of the most important facts in treatment are to hold plasma homocysteine as close as to the normal range [1-8, 21, 23, 24, 26].
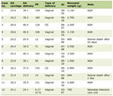 Click to view | Table 1. Different Current and Future Therapeutic Options of HCU |
| Current Treatment Options of HCU | ▴Top |
Supplemental and dietetic aspects
Pyridoxine-responsive cases are treated with pharmacological doses of pyridoxine, vitamin D, and additional administration of folic acid and vitamin B12. Vitamin B6 (pyridoxine) acts as a cofactor for more than 100 enzymes and is involved in many processes of protein and fat metabolism. Cofactor is a kind of “helper molecule” for the enzymes in our body, which in turn are responsible for important processes. The vitamin is essential for the immune system, the nervous system and for the formation of the red blood pigment hemoglobin. In dietary supplements, there is usually the so-called pyridoxine hydrochloride or the already active form pyridoxal phosphate (PLP). Pyridoxine or vitamin B6 is usually present in drugs as pyridoxine hydrochloride (C8H12ClNO3), a white crystalline powder that is readily soluble in water. Pyridoxine is a prodrug that is biotransformed in the organism to the active PLP.
Pyridoxine in its active form PLP is a cofactor of numerous enzymes which play an important role in amino acid, fat and carbohydrate metabolism, among others. The recommended treatment of non-responsive patients is methionine-restricted, cystine-supplemented diet with additional administration of pyridoxine, folic acid, and vitamin B12 [2-9, 11, 17, 19, 21].
Enzyme replacement therapy
The advantage of enzyme replacement therapy is controlled blood levels temporarily in HCU patients. Disadvantages of this therapy includes the life-long therapy with these drugs, and therefore is a very expensive therapy at all. Enzyme replacement therapy includes studies with pegtibatinase (OT-58, TVT-058). The application includes regime twice per week, 1.5 mg/kg subcutaneously [1, 33, 46]. It is a PEG-ylated recombinant enzyme replacement therapy [33, 46]. In 2019, a double blind, randomized, placebo-controlled, phase 1 and phase 2 study to analyze the safety, tolerance, pharmacological aspects pharmacodynamics, and analysis of clinical outcome of pegtibatinase (TVT-058) was initiated [47]. The drug is taken in a subcutaneous application in patients with cystathionine beta synthase deficient HCU (COMPOSE-STUDY) [47]. This study compromising 32 participants will end in 2025 [47]. First results showed a 55% reduction of total homocysteine and a good toleration after 12 weeks [47]. Another enzyme replacement drug, pegtarviliase, is studied in the USA in a phase 1/2 study and is applicated weekly with one dose over 4 weeks. Five cohort studies were performed to date to rule out the efficacy of AEB-4104/pegtarviliase in humans. A phase 1/2 multiple ascending-dosage analysis was initiated in patients with HCU with cystathionine beta synthase deficiency to analyze the safety features, pharmacokinetic function, and pharmacodynamics of pegtarviliase. It is a non-randomized interventional ongoing trial to prove the efficacy in HCU patients [48]. The study is based on two parts: part 1 is an intravenous cohort with four once-weekly (QW) doses of study drug and part 2: three subcutaneous cohorts with four QW doses of study drug [48]. Further studies focus on CDX-6512, a modified human cystathionine gamma lyase. The Food and Drug Administration (FDA) has approved the orphan drug designation (ODD) for CDX-6512 for the treatment of classical HCU in January 2022. CDX-6512 is a methionine-gamma-lyase, which is stable in gastrointestinal tract and is an orally administered enzyme therapy for HCU. CDX-6512 is to date in “pre-IND” development and is an advanced wholly owned program in the company’s biotherapeutic pipeline [52]. Closer information about efficacy in homozygous or severe patients are not known to date; nor there is closer information of what type of patients with HCU can be treated due to preclinical status of research [52]. CDX-6512 is an enzyme developed to be highly resistant to acidic conditions of the stomach and to proteases of the upper intestines. It enables to effectively degrade methionine that is liberated from protein digestion [52]. High levels of this amino acid and its metabolite homocysteine, leads to the various clinical manifestations of HCU [52]. The company presented pre-clinical data from CDX-6512 at the 14th International Congress of Inborn Errors of Metabolism (ICIEM) in November 2021 and showed promising results by poster presentation [52].
Moreover, erymethionase seems to have a significant potential in HCU patients by reducing both levels of homocysteine and methionine in blood plasma, which is critical in restoration of a metabolic balance in patients with HCU [68]. These are, to date, also preclinical studies [68].
| Novel Therapeutic Approaches to Treat HCU | ▴Top |
Chaperones (small ligands)
For some mutant lysosomal enzymes, this thermodynamic instability can be exacerbated, with consequently even less of the properly folded enzyme able to exit the endoplasmic reticulum (ER). Pharmacological chaperones selectively bind and stabilize a specific target enzyme, resulting in increased total cellular levels, and passage through the quality control mechanisms of the ER, with subsequent delivery to lysosomes [28, 65, 66]. Chaperones are proteins that help newly synthesized proteins to fold. The name was chosen after the English term for chaperone, “as they protect immature proteins from harmful contacts”. Newly synthesized proteins must first find their specific, native, functional conformation. This is basically laid out in the primary structure, and smaller proteins can also fold spontaneously in the correct manner. The classic example of spontaneous folding is ribonuclease. However, especially in the case of larger, more complex proteins, aids are often needed to fold correctly, since such proteins tend to form undesirable aggregations that are incapable of functioning. Cells have found a way to minimize aggregation of newly synthesized proteins from the beginning. To do this, the cell makes use of a complex, highly conserved protein machinery called chaperones. These proteins interact specifically with aggregation-prone proteins and thus directly compete with aggregation reactions. The chaperones thereby accelerate the correct folding and association of the proteins without themselves becoming part of the structure. Only non-covalent interactions are affected. The pathogenic mutation in HCU does not target key catalytic rests but introduces structural perturbations leading to a higher tendency of the mutant cystathionine beta synthase to form nonfunctional aggregates and to undergo proteasome-dependent degradation. Correction of the enzyme misfolding represents an alternative therapeutic option for HCU. While roughly half of the patients with HCU respond to a treatment with a PLP precursor pyridoxine, many studies suggested usefulness of small chemicals, such as chemical and pharmacological chaperones or proteasome inhibitors, rescuing mutant cystathionine beta synthase activity in cellular and animal models of HCU. Nonspecific chemical chaperones and proteasome inhibitors assist in mutant cystathionine beta synthase folding process and/or prevent its rapid degradation. The result is an increased steady-state levels of the enzyme and cystathionine beta synthase activity.
Probiotic treatment (SYNB 1353)
Treatment variants for HCU are limited due to efficacy and tolerability. SYNB 1353 is a strain of the probiotic bacteria Escherichia coli (E. coli) type Nissle, which consumes methionine within the gastrointestinal pathway, preventing methionine absorption and conversion to homocysteine in plasma of the patient with HCU. SYNB 1353 is a drug candidate developed to provide a secure, orally applicated, non-systemically absorbed treatment to diminish methionine and to lower homocysteine levels in patients with HCU, thereby lowering the risk of serious side effects [53]. HCU is a metabolic disorder characterized by high levels of homocysteine and risks for thromboembolism, lens disturbances, skeletal deformities, developmental regress, and intellectual disability [53]. It is the first drug candidate developed by a research collaboration study of two biotechnology companies between Synlogic and Ginkgo and the first investigational medicine developed on the platform of Gingko, who were used in clinic. In 2021, Synlogic shared preclinical data for SYNB 1353 that showed significant signs of lowering the blood homocysteine levels in non-human primates and in mouse models [53].
Liver-directed therapy (three dimensional (3D) bioprinting/liver bioengineering)
A bioprinter (bioprinter, organic printer) is a special form of 3D printer, which is computer-controlled to produce regular structures, so-called bioarrays, or tissue from previously grown individual cells using tissue engineering techniques. Later, the technology is expected to make it possible to produce entire organs. Bioprinters could be used in medicine (specific organs), synthetic biology (artificial life forms) and the food industry (artificial meat). Bioprinters are in a very advanced state of development, according to manufacturers. Companies that use bioprinters are Organovo and Modern Meadow. Organovo has medical application plans for the bioprinters, initially to create artificial blood vessels for vascular surgery want to produce synthetic meat as a substitute for industrial animal production. The process is commonly referred to as “bioprinting”. Future research could focus on bioprinting intact liver tissue or liver organoid to transplant into the patient without removing the whole dysfunctional liver.
The liver is a rather complex organ, consisting of different cell types with specific functions, arranged in specific shapes like small “lobules”. Bioengineering of 3D liver constructs is thus challenging but offers great potential for drug discovery/toxic testing as well as disease models.
Hepatocytes will be encapsulated in alginate-methylcellulose (Alg/MC)/Matrigel mixtures for the Shell lane, while fibroblasts were encapsulated in Alg/MC for the Core lane. The fibroblasts acted as supporting cells by stimulating the expression of certain hepatocyte biomarkers, such as albumin. Hepatocytes were shown to proliferate and form multicellular clusters in all conditions. Cluster size, however, was specifically increased in the co-culture experiments with fibroblasts, simultaneously printed in core/shell arrangement. A few feasibility studies considered different bioink compositions (Alg/MC with and without bioactive components), as well as co-cultures vs. monocultures. Different studies were performed in the last years to optimize liver engineered organoids and to make it transplantable in any way into patients with different liver diseases. This could also be an interesting treatment option of HCU patients in the future [75-79].
Liver transplantation
Liver transplantation was performed successfully in six pediatric and also adult patients, whereas one 24 years old man with HCU was cured after liver transplantation and did not receive any drugs or restrictive diet after surgery [39, 54, 55]. In a Chinese population, three vitamin B6 non-responsive cases received liver transplantation at age of 3, 8 and 8 years, respectively [56]. Their blood methionine and total homocysteine returned to normal within a week after liver transplantation [56]. One patient with cholangiocarcinoma and high secondary homocysteine levels received a domino liver transplantation and was alive 1 year after surgery and had low homocysteine levels [55]. It is to discuss, if each HCU patient should receive a liver transplant to refunction the cystathionine beta activity in the patient. Side effects after liver transplantation frequently occur up to graft-versus-host situations. The decision to perform a liver transplantation should be ruled in special severe cases, where other therapy options are not successful.
Gene therapy
Interventions into the genome relate to different approaches preparing a pharmaceutical composition comprising an mRNA stabilized by sequence modifications in the translated region and optimized for translation, or the corresponding modified mRNA. The pharmaceutical composition according to the invention is suitable as a therapeutic agent for gene therapy, in particular for tissue regeneration. Gene therapy and genetic vaccination are molecular medical procedures whose application in the therapy and prevention of diseases will have a significant impact on medical practice. Both methods are based on the introduction of nucleic acids into cells or tissues of the patient and subsequent processing of the information encoded by the introduced nucleic acids, expression of the desired polypeptides. The usual approach of previous methods of gene therapy and genetic vaccination is to use DNA to introduce the required genetic information into the cell. In this regard, various methods for introducing DNA into cells, such as calcium phosphate transfection, polybrene transfection, protoplast fusion, electroporation, microinjection, and lipofection, have been developed, and lipofection in particular has been shown to be a suitable method. Another method that has been proposed, particularly in genetic vaccination procedures, is the use of DNA viruses as DNA vehicles. Such viruses have the advantage that a very high transfection rate can be achieved due to their infectious properties. The viruses used are genetically modified so that no functional infectious particles are formed in the transfected cell. Despite this precaution, however, a certain risk of uncontrolled spread of the introduced gene therapy effective as well as viral genes cannot be excluded due to possible recombination events.
Gene therapy could play an important role in the future to treat HCU in a curable aspect. But at all, gene therapeutical aspects are still in childhood shoes and should urgently further evaluated in detail [37, 57, 58]. Moreover, mutation repair, could play an important role to cure the disease (Table 1) [21, 28, 37, 39, 47, 52-58, 61-79].
| Conclusions | ▴Top |
HCU is a very rare metabolic disease in childhood. Genetic-induced defects in cystathionine beta synthase, a key enzyme of organic sulfur metabolism, results in deficiency of cystathionine beta synthase activity. Current treatment options for classical HCU are very limited and often inefficient, partially due to a low patient compliance with very strict dietary regimen, especially in children. New therapeutical approaches are initiated and are in study to deal with the immense accumulation of homocysteine and to develop a healthy metabolic balance. To date, therapy of HCU patients focus on supplemental and dietary approaches as well as enzyme replacement therapy (Table 1) [21, 28, 39, 47, 52-58, 61-79]. Engineering and chemical modification of human cystathionine beta synthase yielded different enzyme replacement targets like OT-58 or AEB-4104/pegtarviliase, as first-in-class enzyme therapy candidates for HCU. Novel preclinical studies concentrate on targets like CDX-6512 or erymethionase; but to date, it is too early to get any scientific evidence to treat patients with HCU [21, 47]. Enzyme replacement therapies are based on a long-life substitution of a recombinant enzyme that takes the function of the disturbed cystathionine beta synthase in the pediatric patient. “Long-life” means high costs for the health insurance and a good compliance of the parents especially in early years of age in a child with HCU. Treatment with proteasome inhibitors and chaperones could play also an additive role in therapeutical potential due to their potential to bind and stabilize cystathionine beta synthase, resulting in increased total cellular levels after delivery to the lysosomes [28, 65, 66]. Moreover, biotechnical companies in the USA focus in preclinical studies on probiotic treatment with SYNB 1353 [52, 53]. Significant results are, to date, missing [52, 53].
A very interesting aspect curing the disease is based on liver-directed therapy like 3-D-bioprinting and liver bioengineering. Engineering of liver organoids in vitro and later implantation of these tissues into the HCU patient could have a curing aspect in patients with HCU to restore the function of cystathionine beta synthase without completely removing the whole organ in cases of liver transplantation [75, 76, 78, 80-82]. Liver transplantation therefore should only be performed in serious and individual cases, where no other treatment option leads to success. Another aspect is the transplantation of liver progenitor cells from bone marrow for liver regeneration and restoring the function of the disturbed enzyme [71-79]. Cure of patients with HCU could also be achieved by one-stage gene therapy maneuvers in the future. Studies were performed in mice with adeno-associated gene transfer and mini-circle DNA vector gene transfer [37, 56-58, 61, 67, 68, 70].
Further research is necessary in this interesting field of research in this very rare disease in childhood.
Acknowledgments
None to declare.
Financial Disclosure
There is no financial disclosure, nor any funding.
Conflict of Interest
There is no conflict of interest.
Author Contributions
SB performed research, data collection and references; EL and GV read the manuscript and gave important ideas; EM checked grammar and style of the manuscript. LB checked the format and the grammar.
Data Availability
Any inquiries regarding supporting data availability of this review should be directed to corresponding author.
| References | ▴Top |
- Bublil EM, Majtan T. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimie. 2020;173:48-56.
doi pubmed - Tran C, Bonafe L, Nuoffer JM, Rieger J, Berger MM. Adult classical homocystinuria requiring parenteral nutrition: Pitfalls and management. Clin Nutr. 2018;37(4):1114-1120.
doi pubmed - Tankeu AT, Van Winckel G, Campos-Xavier B, Braissant O, Pedro R, Superti-Furga A, Amati F, et al. Classical homocystinuria, is it safe to exercise? Mol Genet Metab Rep. 2021;27:100746.
doi pubmed - Gerrard A, Dawson C. Homocystinuria diagnosis and management: it is not all classical. J Clin Pathol. 2022:75:744-750.
doi pubmed - Khurana DS, Sharma VK, Kaur S, Ram J. Bilateral ectopia lentis in classical homocystinuria. QJM. 2021;114(4):275.
doi pubmed - Saba N, Irshad S. Congenital cataract: An ocular manifestation of classical homocystinuria. Mol Genet Genomic Med. 2021;9(9):e1742.
doi pubmed - Fatima S, Hafeez A, Ijaz A, Asif N, Awan A, Sajid A. Classical homocystinuria in a juvenile patient. J Coll Physicians Surg Pak. 2018;28(6):488-489.
doi pubmed - Rizowy GM, Poloni S, Colonetti K, Donis KC, Dobbler PT, Leistner-Segal S, Roesch LFW, et al. Is the gut microbiota dysbiotic in patients with classical homocystinuria? Biochimie. 2020;173:3-11.
doi pubmed - Poloni S, Siebert M, Donis KC, Weber Hoss GR, Blom HJ, Schwartz IVD. Cytokines levels in late-diagnosed Classical Homocystinuria patients. Mol Genet Metab Rep. 2018;17:43-44.
doi pubmed - Truitt C, Hoff WD, Deole R. Health functionalities of betaine in patients with homocystinuria. Front Nutr. 2021;8:690359.
doi pubmed - Al-Dewik N, Ali A, Mahmoud Y, Shahbeck N, Ali R, Mahmoud L, Al-Mureikhi M, et al. Natural history, with clinical, biochemical, and molecular characterization of classical homocystinuria in the Qatari population. J Inherit Metab Dis. 2019;42(5):818-830.
doi pubmed - Gus PI, Donis KC, Marinho D, Martins TF, de Souza CFM, Carloto RB, Leivas G, et al. Ocular manifestations in classic homocystinuria. Ophthalmic Genet. 2021;42(1):71-74.
doi pubmed - Perry DJ. Hyperhomocysteinaemia. Baillieres Best Pract Res Clin Haematol. 1999;12(3):451-477.
doi pubmed - Martinez-Gutierrez JD, Mencia-Gutierrez E, Gracia-Garcia-Miguel T, Gutierrez-Diaz E, Lopez-Tizon E. Classical familial homocystinuria in an adult presenting as an isolated lens subluxation. Int Ophthalmol. 2011;31(3):227-232.
doi pubmed - El Bashir H, Dekair L, Mahmoud Y, Ben-Omran T. Neurodevelopmental and cognitive outcomes of classical homocystinuria: experience from Qatar. JIMD Rep. 2015;21:89-95.
doi pubmed - Majtan T, Pey AL, Gimenez-Mascarell P, Martinez-Cruz LA, Szabo C, Kozich V, Kraus JP. Potential pharmacological chaperones for cystathionine beta-synthase-deficient homocystinuria. Handb Exp Pharmacol. 2018;245:345-383.
doi pubmed - Allen J, Power B, Abedin A, Purcell O, Knerr I, Monavari A. Plasma methionine concentrations and incidence of hypermethioninemic encephalopathy during infancy in a large cohort of 36 patients with classical homocystinuria in the Republic of Ireland. JIMD Rep. 2019;47(1):41-46.
doi pubmed - Alsharhan H, Ahmed AA, Ali NM, Alahmad A, Albash B, Elshafie RM, Alkanderi S, et al. Early diagnosis of classic homocystinuria in Kuwait through newborn screening: a 6-year experience. Int J Neonatal Screen. 2021;7(3):56.
doi pubmed - Schiff M, Blom HJ. Treatment of inherited homocystinurias. Neuropediatrics. 2012;43(6):295-304.
doi pubmed - Li DX, Li XY, Dong H, Liu YP, Ding Y, Song JQ, Jin Y, et al. Eight novel mutations of CBS gene in nine Chinese patients with classical homocystinuria. World J Pediatr. 2018;14(2):197-203.
doi pubmed - Majtan T, Park I, Carrillo RS, Bublil EM, Kraus JP. Engineering and characterization of an enzyme replacement therapy for classical homocystinuria. Biomacromolecules. 2017;18(6):1747-1761.
doi pubmed - Poloni S, Leistner-Segal S, Bandeira IC, D'Almeida V, de Souza CF, Spritzer PM, Castro K, et al. Body composition in patients with classical homocystinuria: body mass relates to homocysteine and choline metabolism. Gene. 2014;546(2):443-447.
doi pubmed - Ismail HM, Krishnamoorthy N, Al-Dewik N, Zayed H, Mohamed NA, Giacomo VD, Gupta S, et al. In silico and in vivo models for Qatari-specific classical homocystinuria as basis for development of novel therapies. Hum Mutat. 2019;40(2):230-240.
doi pubmed - Okun JG, Gan-Schreier H, Ben-Omran T, Schmidt KV, Fang-Hoffmann J, Gramer G, Abdoh G, et al. Newborn Screening for Vitamin B(6) Non-responsive Classical Homocystinuria: Systematical Evaluation of a Two-Tier Strategy. JIMD Rep. 2017;32:87-94.
doi pubmed - Yi C, He J, Xu J, Zhang X, Huang J. Homocystinuria in a family with novel cystathionine beta synthase gene mutations. Clin Lab. 2021;67(2).
doi - Kruger WD. Cystathionine beta-synthase deficiency: of mice and men. Mol Genet Metab. 2017;121(3):199-205.
doi pubmed - Elsaid MF, Bener A, Lindner M, Alzyoud M, Shahbek N, Abdelrahman MO, Abdoh G, et al. Are heterocygotes for classical homocystinuria at risk of vitamin B12 and folic acid deficiency? Mol Genet Metab. 2007;92(1-2):100-103.
doi pubmed - Majtan T, Pey AL, Ereno-Orbea J, Martinez-Cruz LA, Kraus JP. Targeting Cystathionine Beta-Synthase Misfolding in Homocystinuria by Small Ligands: State of the Art and Future Directions. Curr Drug Targets. 2016;17(13):1455-1470.
doi pubmed - CBS mutations are good predictors for B6-responsiveness: A study based on the analysis of 35 Brazilian Classical Homocystinuria patients. Mol Genet Genomic Med. 2018;6(5):861.
doi pubmed - Lu YH, Cheng LM, Huang YH, Lo MY, Wu TJ, Lin HY, Hsu TR, et al. Heterozygous carriers of classical homocystinuria tend to have higher fasting serum homocysteine concentrations than non-carriers in the presence of folate deficiency. Clin Nutr. 2015;34(6):1155-1158.
doi pubmed - Poloni S, Sperb-Ludwig F, Borsatto T, Weber Hoss G, Doriqui MJR, Embirucu EK, Boa-Sorte N, et al. CBS mutations are good predictors for B6-responsiveness: A study based on the analysis of 35 Brazilian Classical Homocystinuria patients. Mol Genet Genomic Med. 2018;6(2):160-170.
doi pubmed - Majtan T, Jones W, Jr., Krijt J, Park I, Kruger WD, Kozich V, Bassnett S, et al. Enzyme replacement therapy ameliorates multiple symptoms of murine homocystinuria. Mol Ther. 2018;26(3):834-844.
doi pubmed - Park I, Bublil EM, Glavin F, Majtan T. Interplay of enzyme therapy and dietary management of murine homocystinuria. Nutrients. 2020;12(9):2895.
doi pubmed - Maclean KN, Jiang H, Aivazidis S, Kim E, Shearn CT, Harris PS, Petersen DR, et al. Taurine treatment prevents derangement of the hepatic gamma-glutamyl cycle and methylglyoxal metabolism in a mouse model of classical homocystinuria: regulatory crosstalk between thiol and sulfinic acid metabolism. FASEB J. 2018;32(3):1265-1280.
doi pubmed - Levy HL. Early Development of Newborn Screening for HCU and Current Challenges. Int J Neonatal Screen. 2021;7(4):67.
doi pubmed - Hart C, McNulty J, Cotter M, Al Jasmi F, Crushell E, Monavari AA. The challenges of pregnancy management in pyridoxine nonresponsive homocystinuria: The Irish experience. JIMD Rep. 2021;61(1):34-41.
doi pubmed - Sun S, Weile J, Verby M, Wu Y, Wang Y, Cote AG, Fotiadou I, et al. A proactive genotype-to-patient-phenotype map for cystathionine beta-synthase. Genome Med. 2020;12(1):13.
doi pubmed - Lawson-Yuen A, Levy HL. The use of betaine in the treatment of elevated homocysteine. Mol Genet Metab. 2006;88(3):201-207.
doi pubmed - Kerkvliet SP, Rheault MN, Berry SA. Liver transplant as a curative treatment in a pediatric patient with classic homocystinuria: A case report. Am J Med Genet A. 2021;185(4):1247-1250.
doi pubmed - Oliveira Santos M, Geraldes R, Conceicao I. Peripheral nerve involvement in classic homocystinuria: an unusual association. BMJ Case Rep. 2016;2016:bcr2016216255.
doi pubmed - Sellos-Moura M, Glavin F, Lapidus D, Evans KA, Palmer L, Irwin DE. Estimated prevalence of moderate to severely elevated total homocysteine levels in the United States: A missed opportunity for diagnosis of homocystinuria? Mol Genet Metab. 2020;130(1):36-40.
doi pubmed - Valayannopoulos V, Schiff M, Guffon N, Nadjar Y, Garcia-Cazorla A, Martinez-Pardo Casanova M, Cano A, et al. Betaine anhydrous in homocystinuria: results from the RoCH registry. Orphanet J Rare Dis. 2019;14(1):66.
doi pubmed - Betaine anhydrous: new drug. Homocystinuria: continued evaluation needed. Prescrire Int. 2009;18(102):162.
- Tan CRC, Abdul-Majeed S, Cael B, Barta SK. Clinical pharmacokinetics and pharmacodynamics of bortezomib. Clin Pharmacokinet. 2019;58(2):157-168.
doi pubmed - Fricker LD. Proteasome inhibitor drugs. Annu Rev Pharmacol Toxicol. 2020;60:457-476.
doi pubmed - Park I, Hulkova H, Krijt J, Kozich V, Bublil EM, Majtan T. Long-term uninterrupted enzyme replacement therapy prevents liver disease in murine model of severe homocystinuria. Hum Mutat. 2020;41(9):1662-1670.
doi pubmed - Pegtibatinase as an enzyme therapy for patients with homocystinuria caused by cystathionine beta-synthase deficiency (COMPOSE); Interventional clinical trial in 32 participants: A double blind, randomized, placebo-controlled, Phase 1/2 Study to assess the safety, tolerability, pharmacokinetics, pharmacodynamics, and effects on clinical outcomes of pegtibatinase (TVT-058), administered subcutaneously in patients with cystathionine beta-synthase deficient homocystinuria (COMPOSE). ClinicalTrials.gov Identifier: NCT03406611.
- A multiple ascending dose study of ACN00177 (Pegtarviliase) in subjects with CBS deficiency; A phase 1/2 multiple ascending-dose study in subjects with homocystinuria due to cystathionine β-synthase (CBS) deficiency to investigate the safety, pharmacokinetics, and pharmacodynamics of ACN00177. ClinicalTrials.gov Identifier: NCT05154890.
- Yamada K, Yokoyama K, Aoki K, Taketani T, Yamaguchi S. Long-Term Outcomes of Adult Patients with Homocystinuria before and after Newborn Screening. Int J Neonatal Screen. 2020;6(3):60.
doi pubmed - Naughten ER, Yap S, Mayne PD. Newborn screening for homocystinuria: Irish and world experience. Eur J Pediatr. 1998;157(Suppl 2):S84-87.
doi pubmed - Weber Hoss GR, Sperb-Ludwig F, Schwartz IVD, Blom HJ. Classical homocystinuria: A common inborn error of metabolism? An epidemiological study based on genetic databases. Mol Genet Genomic Med. 2020;8(6):e1214.
doi pubmed - Poster, development of an investigational methionine-consuming synthetic biotic medicine (SYNB1353) for the treatment of homocystinuria. International Congress of Inborn Errors of Metabolism. 2021. Nov 21 - 24, 2021 at Hilton Sydney, Sydney, New South Wales, Australia.
- Synlogic initiates phase 1 study of SYNB1353 for the treatment of homocystinuria (HCU). July 12, 2022, Information Synlogic, Australia.
- Lin NC, Niu DM, Loong CC, Hsia CY, Tsai HL, Yeh YC, Tsou MY, et al. Liver transplantation for a patient with homocystinuria. Pediatr Transplant. 2012;16(7):E311-314.
doi pubmed - Qu W, Zhu ZJ, Wei L, Sun LY, Liu Y, Zeng ZG. Feasibility of domino liver transplantation from hyperhomocsyteinemia. Clin Res Hepatol Gastroenterol. 2019;43(5):527-532.
doi pubmed - Li DX, Chen ZH, Jin Y, Song JQ, Li MQ, Liu YP, Li XY, et al. [Clinical characteristics and CBS gene analysis of 13 cases with classic homocystinuria]. Zhonghua Er Ke Za Zhi. 2022;60(6):533-538.
- Wasim M, Khan HN, Ayesha H, Iqbal M, Tawab A, Irfan M, Kanhai W, et al. Identification of three novel pathogenic mutations in cystathionine beta-synthase gene of Pakistani intellectually disabled patients. J Pediatr Endocrinol Metab. 2022;35(3):325-332.
- Morris AA, Kozich V, Santra S, Andria G, Ben-Omran TI, Chakrapani AB, Crushell E, et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017;40(1):49-74.
doi pubmed - Rahman M, Sharma M, Aggarwal P, Singla S, Jain N. Homocystinuria and ocular complications - A review. Indian J Ophthalmol. 2022;70(7):2272-2278.
doi pubmed - Pandey SK, Sharma V. Commentary: Ophthalmic manifestations of homocystinuria. Indian J Ophthalmol. 2022;70(7):2278-2279.
doi pubmed - Al-Sadeq DW, Nasrallah GK. The spectrum of mutations of homocystinuria in the MENA region. Genes (Basel). 2020;11(3):330.
doi pubmed - Majtan T, Kozich V, Kruger WD. Recent therapeutic approaches to cystathionine beta-synthase-deficient homocystinuria. Br J Pharmacol. 2023;180(3):264-278.
doi pubmed - Kisselev AF. Site-Specific Proteasome Inhibitors. Biomolecules. 2021;12(1):54.
doi pubmed - Wang J, Fang Y, Fan RA, Kirk CJ. Proteasome Inhibitors and Their Pharmacokinetics, Pharmacodynamics, and Metabolism. Int J Mol Sci. 2021;22(21):11595.
doi pubmed - Bahr T, Katuri J, Liang T, Bai Y. Mitochondrial chaperones in human health and disease. Free Radic Biol Med. 2022;179:363-374.
doi pubmed - Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18(6):345-360.
doi pubmed - Lee HO, Salami CO, Sondhi D, Kaminsky SM, Crystal RG, Kruger WD. Long-term functional correction of cystathionine beta-synthase deficiency in mice by adeno-associated viral gene therapy. J Inherit Metab Dis. 2021;44(6):1382-1392.
doi pubmed - Testing erymethionase in CBS deficient mice. Erytech Pharma, France.
- Lee HO, Gallego-Villar L, Grisch-Chan HM, Haberle J, Thony B, Kruger WD. Treatment of cystathionine beta-synthase deficiency in mice using a minicircle-based naked DNA vector. Hum Gene Ther. 2019;30(9):1093-1100.
doi pubmed - Park ES, Oh HJ, Kruger WD, Jung SC, Lee JS. Recombinant adeno-associated virus mediated gene transfer in a mouse model for homocystinuria. Exp Mol Med. 2006;38(6):652-661.
doi pubmed - Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, Murase N, Boggs SS, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284(5417):1168-1170.
doi pubmed - Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Teperman L, Henegariu O, et al. Liver from bone marrow in humans. Hepatology. 2000;32(1):11-16.
doi pubmed - Alison MR, Poulsom R, Jeffery R, Dhillon AP, Quaglia A, Jacob J, Novelli M, et al. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406(6793):257.
doi pubmed - Theise ND. Liver stem cells: prospects for treatment of inherited and acquired liver diseases. Expert Opin Biol Ther. 2003;3(3):403-408.
doi pubmed - Griffith LG, Wells A, Stolz DB. Engineering liver. Hepatology. 2014;60(4):1426-1434.
doi pubmed - Mirdamadi ES, Kalhori D, Zakeri N, Azarpira N, Solati-Hashjin M. Liver tissue engineering as an emerging alternative for liver disease treatment. Tissue Eng Part B Rev. 2020;26(2):145-163.
doi pubmed - Zheng MH, Ye C, Braddock M, Chen YP. Liver tissue engineering: promises and prospects of new technology. Cytotherapy. 2010;12(3):349-360.
doi pubmed - Nadi A, Moradi L, Ai J, Asadpour S. Stem cells and hydrogels for liver tissue engineering: synergistic cure for liver regeneration. Stem Cell Rev Rep. 2020;16(6):1092-1104.
doi pubmed - Beckwitt CH, Clark AM, Wheeler S, Taylor DL, Stolz DB, Griffith L, Wells A. Liver 'organ on a chip'. Exp Cell Res. 2018;363(1):15-25.
doi pubmed - Palakkan AA, Hay DC, Anil Kumar PR, Kumary TV, Ross JA. Liver tissue engineering and cell sources: issues and challenges. Liver Int. 2013;33(5):666-676.
doi pubmed - Sudo R. Multiscale tissue engineering for liver reconstruction. Organogenesis. 2014;10(2):216-224.
doi pubmed - Meng F, Assiri A, Dhar D, Broering D. Whole liver engineering: A promising approach to develop functional liver surrogates. Liver Int. 2017;37(12):1759-1772.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.