

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 11, Number 6, June 2019, pages 422-427
Polycythemia in Patients With Hereditary Hemochromatosis: Real or Myth?
Samia Asifa, b, c, Madeline Begemanna, b, Shahzad Razaa, b
aSaint Luke’s Cancer Institute, Kansas City, MO 64111, USA
bUniversity of Missouri, Kansas City, MO 64111, USA
cCorresponding Author: Samia Asif, Saint Luke’s Cancer Institute, 4321 Washington St, STE 4000, Kansas City, MO 64111, USA
Manuscript submitted March 11, 2019, accepted April 9, 2019
Short title: Polycythemia in Patients With Hereditary Hemochromatosis
doi: https://doi.org/10.14740/jocmr3816
| Abstract | ▴Top |
Background: Hereditary hemochromatosis (HH) is an autosomal recessive disorder affecting iron metabolism, resulting in iron accumulation in tissue parenchymal cells. Missense mutations result in homozygosity or heterozygosity for substitutions in the HFE gene, with the most common being C282Y and H63D.
Methods: With an aim to evaluate an association between polycythemia and HH, retrospective chart review was performed for 152 patients with known HFE mutations. Parameters reviewed included individual HFE genotypes, gender distribution, hemoglobin (Hgb) and hematocrit (Hct) levels, median ferritin levels and whether or not phlebotomy was required.
Results: Of 152 patients, 96 (63.2%) were men and 56 (36.8%) were women. Median Hgb and Hct were noted to be higher in men compared to women irrespective of HFE status. Mean age was 60.5 years (range 22 - 93 years). Regarding HFE mutation, 44 (28.9%) patients were C282Y/C282Y, 10 (6.6%) were H63D/H63D and 27 (17.8%) had one copy of each mutation. One patient in the study group was H63D/S65C. Median Hgb and Hct were noted to be 15.5 g/dL and 44.9% respectively in C282Y/C282Y subjects, 16.0 g/dL and 47% in H63D/H63D subjects, 15.8 g/dL and 46% in C282Y/H63D subjects, 16g/dL and 47% in those with single C282Y mutation and 16.6g/dL and 48% in those with single H63D mutation. A total of 67.1% subjects received phlebotomy. A total of 21.7% patients in this cohort were active tobacco users and only 8.6% had an established pulmonary diagnosis, including obstructive sleep apnea (OSA) and chronic obstructive pulmonary disease (COPD). Elevated Hgb levels were noted despite absence of an established reason for secondary polycythemia. Anemia was not encountered despite concurrent medical conditions that would usually be associated with anemia, including gastrointestinal bleeding or end-stage renal disease (ESRD).
Conclusions: Elevated Hgb and Hct levels in HH may be secondary to increased iron uptake by erythroid cell precursors in the bone marrow, in setting of increased availability of both transferrin-bound as well as non-transferrin-bound iron (NTBI). Additional studies need to be pursued to explore the association between HFE mutations and secondary polycythemia.
Keywords: Hereditary hemochromatosis; Secondary polycythemia; HFE mutation
| Introduction | ▴Top |
Hereditary hemochromatosis (HH) is an autosomal recessive disorder affecting iron metabolism, ultimately resulting in accumulation of iron in tissue parenchymal cells. At least five different categories of HH are known, each resulting from mutations in different genes, including HFE, transferrin receptor 2 (TfR2), juvenile HJV (genes encoding hemojuvelin) and juvenile HAMP (genes encoding hepcidin) causing hemochromatosis and ferroportin disease with most common form in Caucasian populations with Northern European descent being HFE mutations [1]. HFE gene is located on the short arm of chromosome 6 (6p21.3) and codes for a major histocompatibility class (major histocompatibility complex, MHC) 1 protein that has a cytoplasmic tail, a transmembrane domain and three extracellular subunits [2, 3]. Missense mutations result in homozygosity or heterozygosity for substitutions in the HFE gene, with the more common being C282Y and H63D, though other less frequent mutations such as H65C are also reported [4]. C282Y homozygosity is reported to account for 80-85% of patients with HH and involves substitution of tyrosine in place of cysteine at amino acid position 282 of the protein product of the HFE gene secondary to a G-to-A missense mutation [5]. Substitution of histidine for aspartate at amino acid position 63 (H63D) and of cysteine for serine at amino acid position 65 (S65C) are also identified [6]. HFE mutations result in uninhibited iron absorption, resulting in transferrin saturation and elevated non-transferrin-bound iron (NTBI) levels and subsequently, deposition in various parenchymal cells. If left untreated, HH results in multi-organ dysfunction.
Polycythemia refers to elevated hemoglobin (Hgb) and hematocrit (Hct) levels in peripheral blood; for men, Hgb > 16.5 g/dL and Hct > 49% while for women Hgb > 16 g/dL and Hct > 48% comprise polycythemia [7]. It has been conjectured in few prior studies that HFE HH may be associated with elevated mean Hgb levels [8] though no definite association has yet been established and screening for HH is not recommended in routine workup for secondary polycythemia. In a review of 634 C282Y homozygous patients at the London Health Sciences Center (London Ontario), Beaton et al had reported a mean Hgb level of 145 + 13 g/L [9]. Patients who have hemochromatosis rarely require blood transfusions.
In this study, we review the median Hgb and Hct in patients with HFE mutation in the Midwest United States health system with the aim to better understand prevalence of polycythemia in hemochromatosis patients. We also observed whether a higher nadir of Hgb and Hct was present in patients with HFE mutation irrespective of low to normal ferritin levels and how decision to pursue phlebotomy correlated with Hgb and Hct levels and underlying HFE mutation status.
| Patients and Methods | ▴Top |
The study was approved by Institutional Review Board (IRB) at Saint Luke’s Health System (Kansas City, MO, USA) and was conducted according to ethical principles for research involving human subjects as stated in the Declaration of Helsinki.
We identified patients who were seen in hematology clinic within the Saint Luke’s System and had known HFE mutations. Retrospective chart review was performed to identify individual HFE genotypes and only patients whose exact mutation was known were included. Parameters that were evaluated included gender, age, Hgb, Hct and ferritin levels. Hgb and Hct levels included in data were from time of diagnosis or from time of first visit if previously sought care at outside facility. Also noted was whether phlebotomy was pursued as a treatment option, and associated medical conditions such as obstructive sleep apnea (OSA) or chronic obstructive pulmonary disease (COPD) that could contribute to erythrocytosis as well as conditions like gastrointestinal bleeding or malignancy requiring chemotherapy that could contribute to anemia were noted. Data was grouped into categories based on C282Y or H63D heterozygosity or homozygosity (i.e. C282Y/C282Y, C282/wild type, H63D/H63D, H63D/wild type) or C282Y and H63D heterozygosity (C282Y/H63D). Also included were patients who were noted to be heterozygous for S65c mutation; one patient was noted to be heterozygous for both H63D and S65c and one was heterozygous for C2A2Y. Final data included 160 patients.
| Results | ▴Top |
The study included 160 patients who were referred to hematology for evaluation of HH. Subsequently, eight patients were excluded as described below and data was analyzed for 152 patients. Of these 152 patients, 96 (63.2%) were male and 56 (36.8%) were female. Mean age was 60.5 years (range 22 - 93 years). Of these patients, 44 (28.9%) patients were homozygous for C282Y mutation, 10 (6.6%) were homozygous for H63D and 27 (17.8%) had one copy of each mutation. Only one patient in the study group was heterozygous for H63D and S65C mutations. Table 1 and Figure 1 summarize the frequencies of individual gene mutations observed in the study group.
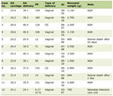 Click to view | Table 1. Patient Characteristics (n = 152) |
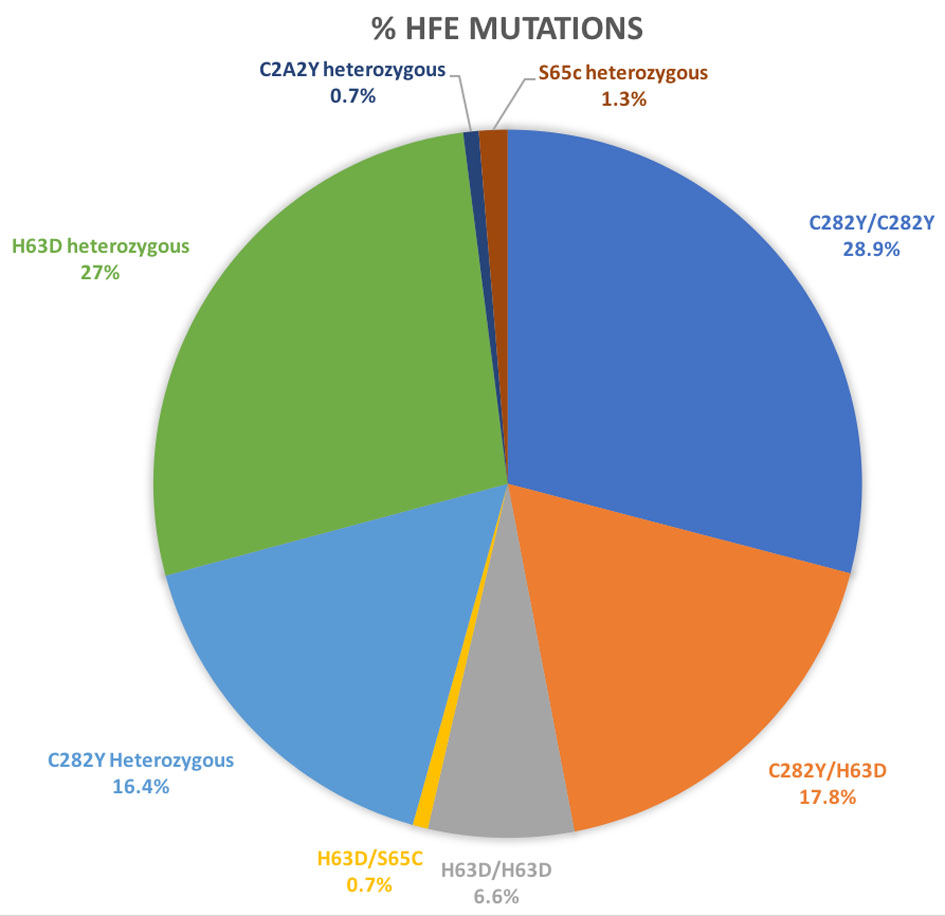 Click for large image | Figure 1. HFE mutations in study participants (n = 160). |
For each HFE mutation cohort, median Hgb, Hct and ferritin levels were identified. Frequency of co-existing conditions that could result in secondary polycythemia was also noted; the most common of these included OSA and active tobacco use. Also identified were some major etiologies that could lead to low Hgb levels; this included receiving chemotherapy for co-existing diagnosis of malignancy, gastrointestinal bleeding and menorrhagia contributing to blood losses and end-stage renal disease (ESRD). Of note, one subject in the study group was found to have JAK V617F mutation and was diagnosed with primary myelofibrosis; this patient was heterozygous for C282Y. Initial data was collected based on 160 consecutive patients; however, with intent to simplify interpretation of results, patients who had received chemotherapy (n = 7) and one patient with primary myelofibrosis were excluded on subsequent analysis, unfortunately with an unavoidable risk of introducing selection bias. Results are reported for 152 patients after exclusion of these eight patients. A total of 11/152 (0.07%) patients had a formal diagnosis of OSA; only one patient was noted as having COPD. A total of 33/152 subjects (21.7%) reported active tobacco use.
For subjects homozygous for C282Y mutation (C282Y/C282Y), median Hgb was noted to be 15.5 g/dL with median Hct of 44.9%. For subjects homozygous for H63D mutation (H63D/H63D), median Hgb was 16g/dL with median Hct of 47%. For individuals heterozygous for single mutation each of C282Y and H63D (C282Y/H63D), median Hgb was noted to be 15.8g/dL with median Hct of 46%. The only patient in the study group with heterozygosity for both H63D and S65C (H63D/S65C) had an Hgb level of 17.5g/dL with an Hct of 49%. For patients with a single HFE mutation, median Hgb and Hct were also noted; results are summarized in Table 1. Median Hgb and Hct were noted to be higher in men compared to women irrespective of HFE status; this is compared in Table 2.
Click to view | Table 2. Comparison of Hemoglobin and Hematocrit Levels Between Men and Women |
Median ferritin in patients who received phlebotomy was higher than that in those not receiving phlebotomy, irrespective of genotype. There were 102 out of 152, namely 67.1% subjects, who received phlebotomy. This included 38/44 (86.4%) subjects with C282Y/C282Y, 20/27 (74.1%) subjects with C282Y/H63D and 6/10 (60%) subjects with H63D/H63D. Of note, patients who were heterozygous for HFE mutation were also treated with phlebotomy, including 15/26 (57.7%) subjects with single C282Y mutation and 19/41 (46.3%) subjects heterozygous for H63D. This suggests that clinical diagnosis of hemochromatosis that requires intervention may be independent of hemochromatosis mutation status. Median ferritin for 102 patients who received phlebotomy was 526 ng/mL; median ferritin value for 50 patients who did not receive phlebotomy was 150.5 ng/mL. Median ferritin levels were noted to be highest in patients with C282Y/C282Y genotype. Median ferritin values for individual genotypes are summarized in Table 3.
Click to view | Table 3. Median Ferritin Levels in Patients Receiving and not Receiving Phlebotomy in Each Subset of HFE Mutations |
Median Hgb for patients who required phlebotomy (n = 102) was 16 g/dL with median Hct of 47%. For those who did not require phlebotomy (n = 50), median Hgb was 16.2 g/dL and median Hct was also 47%. This suggests that while patients who did get phlebotomy did have higher ferritin levels when compared to those who did not get phlebotomy, the Hgb and Hct values were neither affected by serum ferritin levels, nor the decision to pursue phlebotomy.
| Discussion | ▴Top |
The normal Hgb level is 13.5 to 17.5 g/dL for men and 12 to 16 g/dL for women. The normal Hct is 40-54% for men and 36-48% for women [10]. An association between hemochromatosis and erythrocytosis was reported as early as 1958, when McFadzean et al reported a case series in which erythrocytosis was observed in patients with HH in setting of hepatoma [11]. Literature review reveals multiple cases reported since then in which the relation between polycythemia and hemochromatosis was described, but only in presence of hepatoma and attributed to erythropoietin production by the hepatoma. Given improvements in diagnostic testing in current era, hemochromatosis is frequently identified early, prior to development of end organ damage such as liver cirrhosis or hepatocellular cancer, and continued observation of generally higher Hgb levels suggests a more direct relation between HFE mutation and hematological parameters. In our study for participants, who each carried at least one HFE mutation, higher median Hgb levels were noted in most patients. An interesting observation remained that anemia was not encountered in this group of 152 patients with underlying HFE mutation. This was a consistent observation even in absence of high ferritin levels, such as in subjects not deemed to require phlebotomy. Etiology to explain the absence of anemia in this group of patients, particularly those with concurrent medical conditions that would make them more susceptible to lower Hgb levels such as gastrointestinal bleeding or ESRD and those who had already received a phlebotomy at time of first available Hgb level, remains unclear but suggests a correlation between presence of HFE mutation and higher Hgb and Hct levels. While prior studies have indicated elevated Hgb levels in C282Y homozygotes [12], results from our study showed even carrier states for HFE mutation are associated with elevated Hgb and Hct irrespective of serum ferritin value and presence or absence of secondary causes that can cause polycythemia.
Attention was paid to the presence of etiologies that could lead to secondary polycythemia; 21.7% (n = 33/152) patients in this cohort were active tobacco users and only 8.6% had an established pulmonary diagnosis, including OSA and COPD (n = 12/152); one patient had multiple pulmonary embolisms requiring chronic anticoagulation (n = 1/152). Consistently elevated Hgb levels were noted despite absence of an established reason for secondary polycythemia in most subjects. It is to be noted that some patients had received phlebotomies for variable periods of time prior to being seen at Saint Luke’s Cancer Center, as a result of which initially charted Hgb levels may have been falsely low as these patients had been receiving scheduled phlebotomy prior to laboratory testing.
A prior study by Barton et al attempted to investigate the impact of hemochromatosis on peripheral blood parameters including Hgb and Hct levels between patients with HFE mutations and matched normal controls. The study suggested increased values of mean Hgb and Hct in subjects with C282Y/C282Y genotype [13].
Investigating this association further, Franchini et al screened for presence of HFE gene mutation in 52 patients with a pre-existing diagnosis of polycythemia vera (PV). Twelve patients were found to have HFE mutations, two being H63D/H63D whereas the remaining heterozygous for either C282Y or H63D. No patients were noted to be homozygous for C282Y mutation. The study concluded no increase in prevalence of HH in patients with PV [14].
Understanding the link between HFE gene mutation and Hgb levels remains crucial, because HH is a commonly encountered inherited disorder and if an association does exist, the risk of not recognizing a common etiology for elevated hematological parameters arises. This may be deleterious in multiple ways, such as resulting in expensive unnecessary workup, such as investigating JAK2 mutations for evaluation of myeloproliferative disorders in a patient who may have erythrocytosis by virtue of underlying HFE mutation as well as undue anxiety for patients.
Iron in circulation is bound to heme, ferritin and transferrin. Transferrin transports iron in the plasma and delivers iron to bone marrow erythroid cells. Mechanism for elevated Hgb levels in HH can be speculated to be secondary to elevated transferrin saturation levels, which may cause increased iron uptake by erythroid cells resulting in increased red blood cell production. Presence of NTBI has been reported in conditions associated with chronic iron overload with elevated transferrin saturation such as HH; noted to be present in plasma in HH even when transferrin saturation levels are normal and may be used by erythroid cells by a transferring-independent pathway [15]. Our study however notes elevated median Hgb and Hct values irrespective of corresponding median ferritin levels, suggesting possible presence of alternate pathways to explain these higher Hgb and Hct levels in patients with HFE mutations that are yet to be elucidated. Further investigation and study are required to delineate exact underlying mechanisms.
Future direction
Iron homeostasis is a complex process involving multiple cellular pathways. Hepcidin, a liver synthesized peptide, binds to ferroportin, the transmembrane iron-transport channel between cells involved in iron export (such as macrophages, duodenal enterocytes and hepatocytes) and the plasma. Hepcidin binding results in endocytosis and degradation of ferroportin, inhibiting iron transport from cell to the plasma. Hepcidin synthesis is controlled by plasma iron levels and body iron stores in a negative feedback manner which involves regulatory ligands such as hemojuvelin (HJV), transferrin receptors 1 and 2 and HFE transmembrane protein [16, 17]. In HH, genetic mutations may involve the hepcidin gene itself or may result in disruption of regulatory mechanisms, leading to reduced hepcidin production. Alternatively, ferroportin may become resistant to hepcidin modulation.
Development of drugs targeting the hepcidin-ferroportin axis is a potential area for future research that will allow additional treatment options apart from phlebotomy and chelation therapy to be made available to patients with HH. Hepcidin agonists, either agents with endogenous hepcidin activity or those that stimulate production of hepcidin by hepatocytes, have been studied in animal models [18]. This also includes regulatory proteins involved in iron metabolism pathways, such as matriptase 2 (TMPRSS6), a protease that downregulates hepcidin production [19]. Several hepcidin agonists, including hepcidin mimetics, ferroportin inhibitors and stimulators of hepcidin production, are in clinical trials [20].
Conclusions
Presence of HFE mutation is associated with a lower prevalence of anemia and elevated Hgb and Hct levels. This may be secondary to augmented iron uptake by erythroid cell precursors in the bone marrow in setting of iron transport by increased transferrin-bound as well as NTBI. The association is present in subjects both homozygous for as well as carriers of HFE mutations. Elevated levels of Hgb and Hct were noted even in absence of variables that are known to cause secondary polycythemia. Interestingly, despite presence of features such as menorrhagia and gastrointestinal bleeding, significant anemia was not documented in this group of patients. Additional studies need to be pursued to explore the association between HFE mutations and Hgb and Hct levels.
Acknowledgments
None to declare.
Financial Disclosure
No funding was received.
Conflict of Interest
We do not have financial and non-financial competing interest. No writing assistance was utilized in the production of the manuscript.
Informed Consent
Waiver of informed consent process was approved by IRB given a retrospective chart analysis was performed and no patient identifiers were included during data collection and analysis.
Author Contributions
SA and SR designed the study and drafted the manuscript. SA collected retrospective data. MB reviewed the manuscript. SR supervised the entire project and reviewed the manuscript. All the authors have read and approved the final version of this manuscript.
| References | ▴Top |
- McLaren GD, Gordeuk VR. Hereditary hemochromatosis: insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematology Am Soc Hematol Educ Program. 2009:195-206.
doi - Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399-408.
doi pubmed - Burke W, Thomson E, Khoury MJ, McDonnell SM, Press N, Adams PC, Barton JC, et al. Hereditary hemochromatosis: gene discovery and its implications for population-based screening. JAMA. 1998;280(2):172-178.
doi pubmed - Beutler E, Gelbart T, West C, Lee P, Adams M, Blackstone R, Pockros P, et al. Mutation analysis in hereditary hemochromatosis. Blood Cells Mol Dis. 1996;22(2):187-194; discussion 194a-194b.
doi - European Association For The Study Of The L. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3-22.
doi - Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, American Association for the Study of Liver D. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328-343.
doi pubmed - Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer (IARC): Lyon; 2017.
- Beutler E, Felitti V, Gelbart T, Waalen J. Haematological effects of the C282Y HFE mutation in homozygous and heterozygous states among subjects of northern and southern European ancestry. Br J Haematol. 2003;120(5):887-893.
doi pubmed - Beaton MD, Adams PC. The myths and realities of hemochromatosis. Can J Gastroenterol. 2007;21(2):101-104.
doi - Billett HH. Hemoglobin and Hematocrit. In: Walker HK, Hall WD, Hurst JW, eds. Clinical methods: the history, physical, and laboratory examinations. 3rd edition. Boston: Butterworths, 1990: Chapter 151. Available from: https://www.ncbi.nlm.nih.gov/books/NBK259/.
- Mc FA, Todd D, Tsang KC. Polycythemia in primary carcinoma of the liver. Blood. 1958;13(5):427-435.
- Beutler E, Felitti V, Gelbart T, Ho N. The effect of HFE genotypes on measurements of iron overload in patients attending a health appraisal clinic. Ann Intern Med. 2000;133(5):329-337.
doi pubmed - Barton JC, Bertoli LF, Rothenberg BE. Peripheral blood erythrocyte parameters in hemochromatosis: evidence for increased erythrocyte hemoglobin content. J Lab Clin Med. 2000;135(1):96-104.
doi - Franchini M, de Matteis G, Federici F, Solero P, Veneri D. Analysis of hemochromatosis gene mutations in 52 consecutive patients with polycythemia vera. Hematology. 2004;9(5-6):413-414.
doi - Brissot P, Ropert M, Le Lan C, Loreal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820(3):403-410.
doi pubmed - Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531-539.
doi pubmed - Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281(39):28494-28498.
doi pubmed - Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:538-542.
doi pubmed - Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, Hettinger J, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121(7):1200-1208.
doi pubmed - Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018;131(16):1790-1794.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.