

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 10, Number 12, December 2018, pages 857-867
Biovigilance for the Quality and Safety of Medical Products of Human Origin
Vasiliki Gkiokaa, h, Panagiotis Tsirigotisb, Markos Sarrisc, Sotiris Soulisc, Athanasios Apostoloud, Michel Noutsiase, Georgios Dimitriadisb, Alkiviadis Kostakisa, Ioannis Boletisf, Andreas Karabinisg
aBiomedical Research Foundation Academy of Athens, Athens, Greece
bSecond Propedeutic Department of Internal Medicine, “Attiko” University General Hospital, National and Kapodistrian University of Athens, Athens, Greece
cHealth and Social Care Management, University of West Attica, Athens, Greece
dDepartment of Immunology and Histocompatibility, School of Health Sciences, Faculty of Medicine, University of Thessaly, 41500, Larissa, Greece
eDivision of Cardiology, Department of Internal Medicine III, Mid-German Heart Center, Angiology and Intensive Medical Care, University Hospital Halle, Martin-Luther-University Halle-Wittenberg, Ernst-Grube-Strasse 40, D-06120 Halle (Saale), Germany
fNephrology Clinic and Kidney Transplant Unit, “Laiko” General Hospital, School of Medicine, National and Kapodistrian University of Athens, Athens, Greece
gThe Onassis Cardiac Surgery Center, Cardiac Surgery Intensive Care Unit, National and Kapodistrian University of Athens, Athens, Greece
hCorresponding Author: Vasiliki Gkioka, Hellenic Cord Blood Bank, Biomedical Research Foundation Academy of Athens (BRFAA), Athens, Greece
Manuscript submitted July 28, 2018, accepted August 13, 2018
Short title: Biovigilance for Human Medical Products
doi: https://doi.org/10.14740/jocmr3549w
- Abstract
- Introduction
- Risks, Adverse Events and Reactions
- Reporting Adverse Reactions at the Donor
- Reporting Adverse Events at the Recipient
- Adverse Events Report
- The Biovigilance System
- Management of SAR and SAE
- Conclusions
- References
| Abstract | ▴Top |
Progress in science and technology in the health services has led to the development of methods of regenerating and replacing solid organs, tissues and cells, using human body components to create medical products of human origin intended for clinical use. In the activities in which products of human origin are used, however, from the point of donation and harvesting to the subsequent care of the recipient, medical products of human origin are exposed to the risk of specific complications related to the transmission of infectious diseases, and further side-effects. Biovigilance system application is a basic requirement for ensuring the quality and safety of tissues and cells intended for human use. The quality system focuses on error prevention, maintaining a consistent pattern of agreed assays for tissues and cells intended for clinical use. The implementation of quality and safety standards, the development of medical protocols and cooperation protocols between member states, the implementation of Single European Code (SEC), and the development of electronic traceability systems, all aim at vigilance and the surveillance of medical products of human origin from donation to transplantation.
Keywords: Assurance; Biovigilance; Cells; Safety; Serious adverse event; Serious adverse reaction; Tissues
| Introduction | ▴Top |
Progress in science and technology in health services has fostered the development of methods of regenerating and replacing solid organs, tissues and cells, using human body components to create medical products of human origin (MPHO), intended for clinical use [1]. The word “product” refers to human body components which are suitable for clinical use and are derived from specific processes that combine human labor with technological intervention [2].
More than 30,000,000 people in Europe receive MPHO annually, representing on the one hand “traditional products” whose therapeutic interest has been recognized for decades (blood, solid organs, primary hematopoietic stem cells, reproductive cells, tissues, breast milk, etc.), and on the other hand biotechnology products (e.g. decellularized vessels, valves etc.) as well as scaffolds in full development [3].
MPHO often represent the most beneficial and cost-effective treatments for several life-threatening diseases and differ fundamentally from other medical products, because the only possibility of finding them results from the offer of living or cadaveric donor. For this reason, high ethical standards are required in management of relative products considering the right of the donor in health and assuring that it is not subject to any exploitation, coercion or abuse [4].
Each year, 20 million blood donations are held in the European Union (EU) that are distributed from 1,300 blood donations, allowing about 26 million transfusions of patients. One million tissues and cell donations also take place, which are managed by > 3,000 tissue establishments annually. Many member states exchange transnational MPHOs and costs which arise only for blood, tissues and cells account for about 6 billion€ annually, while healthcare services created cost significantly multi fold amount. The organization and management of hematopoietic stem cells transplantation arises an additional cost of 3 billion € annually [5]. MPHO are also used as raw materials for pharmaceutical products, such as plasma derivatives and advanced therapy medicinal products (ATMP). In particular, plasma is important for the production of drugs that are estimated having an annual market value of 4 billion € in the EU.
Despite all the above, the inadequate donation rates, particularly of organs, and the lack of MPHO is of great concern and varies according to the developmental level of each country. Despite estimates, > 150,000 corneal tissue grafts at worldwide scale do not satisfy the annual demand and this is a major concern in many countries from India to Canada [6, 7]. Also, knowledge is limited regarding gametes shortcomings. In China [8], potential recipients can expect sperm for 2 years or up to 6 years for ovules in France [9].
| Risks, Adverse Events and Reactions | ▴Top |
Like all activities for which products of human origin are used, from donation to the subsequent care of the recipient, the MPHO are exposed to risks of specific complications, which are mainly related to the transmission of infectious diseases and other adverse events. It is estimated that a donor has an average of five MPHO, which are subsequently processed individually, creating a network with many branches until their transplantation to recipient(s) [10].
A vigilance and surveillance program is a key prerequisite for tissue and cell quality and safety assurance during application to humans. The quality system focuses on preventing mistakes and errors, while maintaining a standard pattern of agreed definitions for tissues and cells intended for clinical use. However, occasionally, residual risks or errors result in failures, transmitted diseases or conditions in which donors or patients/recipients are exposed to risks, even if not being damaged.
According to the EU definitions, a serious adverse event (SAE) in the present conceptual designation is any undesirable incident related to sampling, testing, processing and distribution of tissues and cells, which could lead to transmission of an infectious disease, death or life-threatening disease, disability or incapacity of the patient or which could lead to prolonged hospitalization or increased morbidity. Also, the 2004/23/EC [11] directive defines the “serious adverse reaction” (SAR) as an “unintended” reaction, including donor or recipient infectious disease, associated with the procurement or application to human tissues and cells, which is fatal, life-threatening, causing disability or incapability or leading to prolonged hospitalization or morbidity”.
According to the above definition, equal importance is given to both donors and recipients. Hence, serious reactions at donors associated with the procurement should be reported. The adverse events are categorized as follows: 1) SAR: damage to the donor, recipient, embryo or offspring; 2) SAE: risk of damage. These definitions are also accepted by the World Health Organization (WHO) [10].
As determined by the definitions these two types of adverse events, an unfortunate incident that has led to severe damage to a donor or recipient is reported as SAR, while an unfortunate incident which has compromised but does not have, or does not already have, caused damage, is referred to as SAE. It should be noted that only one report is disclosed for each incident. Even when a SAR is the result of an adverse event, once a recipient or a donor has suffered damage it prevails, and the incident is reported as SAR. The only exception is when an adverse event leads to a donor SAR reaction and when that particular reaction does not fall within the mandatory reporting criteria (i.e., a defect in quality or safety is not caused or occurs in donated tissues or cells).
For example, a hematopoietic stem cell donor presents an SAR during the collection that should be excluded due to known health risk factors. However, the cells are suitable for transplantation. In this case, the donor’s SAR may be reported as a non-mandatory reaction as there is no impact on the quality or safety of cells although SAR (the error in donor selection) can also be reported as it meets the criteria of mandatory report. In this case, it is recommended that the event should be reported as mandatory SAR and there action should be reported to the non-mandatory category of donor reactions, if this procedure is applied in a particular Member State.
Both the procurement organizations (Pos) and organizations responsible for human application (ORHA), in cooperation with the CA, should promote a reporting and notifying profile (report submission) of SAE and reactions (SAR), because the ability of learning and improvement is provided, and it should not be related to guilt or punishment. Similarly, identifying and submitting reports for suspicious SARs requires awareness of health care professionals of the potential consequences that may have for others. Clinicians should be encouraged to be alert for clinical situations that could potentially be caused by cells and tissues and adverse reactions that should be monitored with extreme caution.
The minimum requirements described in Article 5 of 2006/86/EC [12] directive require the POs notify tissue establishments (TEs) about the SARs of donors only when it affects the quality or safety of tissues and cells. Nevertheless, this approach is not consistent with the recital of Article 9 of the same directive, which states that: “Serious adverse reactions can be detected during or after procurement of living donors or during or after transplantation to humans. Serious adverse reactions shall be reported to the partner tissue establishment for subsequent investigation and notification to the competent authority”.
In conclusion, an adverse reaction is an incident in which a living donor, a recipient or an embryo or a child resulting from in vitro fertilization or intrauterine insemination (IUI) with gametes donors has been damaged; while an adverse event is an incident that causes risk of damage, though no actual damage can occur. All adverse events and reactions should be documented by health care professionals to ensure the appropriate investigation, as well as the corrective and preventative actions. Those incidents classified as “severe” should be notified to competent authorities according to national law. Although adverse events may arise from procurement to the distribution of tissues and cells, most of them are not “severe” and could be possibly managed through the Quality Management System (QMS) of the tissue establishment. On the contrary, SAR and SAE are relatively rare. Consequently, there are notable benefits of data integration at regional, national or international level in practical terms [13].
| Reporting Adverse Reactions at the Donor | ▴Top |
There are several types of living tissue and cells donors. Generally, they can be categorized as autologous and allogeneic donors. The largest group is represented by living donors of hematopoietic stem cells: bone marrow, peripheral blood or umbilical cord blood cells. Allogeneic donors could be related to the recipient (this case is referred as “directed donation”) or not, in this case they are called adult volunteers donor and umbilical cord blood units.
Donation of tissues from living donors is usually associated with the removal of tissue for unrelated reasons to the use in another patient (sometimes referred to as “surgical residue”). Relative examples are bone donation during primary hip replacement or skin donation after removal for aesthetic reasons. In these cases, the risks for the patients/recipients of the tissue graft are usually associated to surgery itself rather with donation. A bone can also be removed from patients for autologous use, and in some cases, the respective processes can be associated to adverse effects or complications.
It is noted that through continuous follow-up of the living donors a reaction can be determined and assured, which is not known during donation but appears in the donor later and may have an impact on the recipient. In these cases, there should be a documented procedure to notify the recipient transplant center immediately for that particular situation [14].
For example, the best donor follow-up practice after hematopoietic stem cells donation should include the following: 1) Specific questionnaire for the collection of information on physical and psycho-emotional condition of the donor; 2) Medical interview in case of pathological conditions that occur after the donation and are possibly related to it; 3) Physical examination in every case considered necessary; 4) Blood test including at least the platelet count; 5) Diagnostic tests in case of further clinical information.
Therefore, all adverse events and reactions which are suspected of being associated with the quality and safety of tissues and cells should be notified to tissue establishments by the clinical users and organizations that conduct the removal of tissue or cellular graft from living donors, so that the trend of all minor events and reactions can be monitored, for the reason of continuous improvement. The tissue establishments should then identify these SAR, which should be notified to the competent authority (CA). EU directives, with great precision, require that donor reactions should only be reported in case the quality and safety of tissues has been affected, while other states require, or at least accept, reporting any donor reactions, irrespective of whether the quality and safety of tissues or cells have been affected or not.
In the context of the SOHO vigilance and surveillance (V&S) [15] project issues were addressed such as the association between SAE with tissues and cells donation, as well as whether their transplantation to humans has caused events that can be evaluated and investigated. In the same program project research was conducted by the Spanish Agency of Transplantation that included a question regarding report submission requirements for SAR to donors, even if quality and safety of the above mentioned tissues and cells had not been affected. Twenty-eight national CA from 26 EU member states responded (in two cases there were different CAs in the field of Medically Assisted Reproduction Units (MARU)). Nineteen of the 28 CA reported requiring the report of the SAR of the donor, even at said conditions, while nine reported not requiring the relative report. The types of donor reactions for which reports were usually required were reactions associated with ovarian hyperstimulation in MARU, with granulocyte colony stimulating treatment agent for the collection of peripheral blood hematopoietic stem cells and reactions (i.e. calcium toxicity), also during collection of peripheral blood stem cells [14] (Fig. 1).
 Click for large image | Figure 1. Adverse reactions at the donor requiring report in the member states of the European Union. Source: ARTHIQS [14]. GCSF AT: Reactions due to GCSF in autologous patients; GCSF AL: Reactions due to GCSF responses at allogeneic donors; PBSC AT: Toxicity during peripheral blood collection (PBSC) in autologous donors; PBSC AL: Toxicity during peripheral blood collection (PBSC) in allogeneic donors. |
Other reactions to the donor requiring reporting are described in general terms as any reactions resulting in damage, medical intervention or hospitalization of the donor. The survey reported that less than one third of the member states maintained records of the living donors, so their health is monitored in long-term after donation. In most cases, the respective records are kept at central level (national registries) and mainly concern donors of primary hematopoietic stem cells. Some member states report developing registries for donors of reproductive cells.
The result of the above survey led the European Commission (EC) to formulate/issue guidelines for member states in 2009 and in 2010 for the completion of the Annual Report of Serious Adverse Reactions and Events. On these guidelines, which are referred to as “document of common approach”, it is stated that “The Commission recognizes the value of these data (i.e. reports of donor reactions, which have not affected the quality and safety of tissues and cells), in the context of tissue and cell regulation and invites member states to submit an annual report regarding the donor’s reactions reported to the CA on a voluntary basis”. An additional non-mandatory category was also introduced to the electronic reporting system regarding donor’s reactions that do not affect quality and safety of tissues and cells, reported data which will not be counted as part of the total number of SAR.
This report aimed at summarizing the type of adverse events that occur more often in donors of tissues and cells, in order to examine whether they meet the criterion of affecting the quality and safety of tissues or cells and to formulate/issue recommendations on good practice regarding the submitting reports of these reactions in the EU.
| Reporting Adverse Events at the Recipient | ▴Top |
The 2004/23/EU directive, article 11, paragraph 1, states in particular that “member states shall provide-ensure that there is a system for reporting, recording and transmitting information regarding serious adverse events and reactions, which could affect the quality and safety of tissues and cells and which may be due to procurement, testing, processing, storage and distribution of tissues and cells, as well as any another serious adverse reaction found out during or after clinical application, which is may be associated with the quality and safety of tissues and cells”.
A reaction from the application of a tissue or cell can occur at any time post-transplantation. There should be a causal relationship with the transplantation or other clinical application. Adverse reactions should be reported, investigated and evaluated in terms of severity, allocation, probability of repetition or frequency, as well as consequences [16]. For this reason, the competent institutions should have effective systems of rapid isolation or recall of unsafe tissues or cells concurrently with traceability procedures in order to determine the time point when the recipient was compromised. A notified reaction may become an essential learning tool for health care professionals.
An effective system of V&S is largely relying on every health care professional involved, from the procurement to clinical application, and in particular on: 1) The medical staff (including surgeons) involved in tissues and cells procurement activities, who during follow-up of the living donors can be informed about further details on the safety of said graft; 2) The staff conducting procurement of tissues and cells per se; 3) The clinical users, who should be alert for adverse effects and be able to know when these effects could be associated with the tissues or cells transplanted; 4) The doctors who take care of children born by assisted reproduction method without partner and could possibly identify a genetic anomaly, with the report of which to prevent further distribution of gametes/embryos from said donor; 5) Any other staff of the tissue establishment, who is involved in either the donation/removal activity or the transplantation activity of tissues and cells; 6) Other vigilance systems (e.g. hemovigilance, pharmacovigilance), when issues are identified that may affect the safety of transplantation tissues or cells.
Adverse reactions may occur from several factors associated with surgery or the condition of the patient. Therefore, clinicians may not consider tissues or cells implemented as a potential source of the adverse event. For this reason, the tissue establishments procuring tissues and cells should encourage tissue and cell procurement and transplantation organizations to consider whether an adverse event can be associated with the donation process or caused by the tissues or the cells transplanted, so that similar future reactions can be avoided [17].
Determination of adverse reactions
Several symptoms or situations indicate that an adverse reaction could occur to a recipient of a tissue or cell, and therefore should be considered as a “trigger” for reporting the adverse reaction. For report submission in the EU, there is a requirement from the CAs to report the following reactions: 1) Transmissible bacterial infection; 2) Transmissible viral infection; 3) Transmissible parasitic infection, transmissible malignant diseases, other transmissible diseases; 4) Others adverse events.
Clinicians should investigate for symptoms or conditions suggesting that any of the following reactions may have occurred due to the transplantation of the product to the recipient: 1) Infection from the donor: unexpected primary infections possibly transmitted from the donor to the recipient (e.g. viral, bacterial, parasitic, fungal, prion); 2) Infection from tissues/cells: transmissible disease (viral, bacterial, parasitic, fungal, prion) likely due to infection or cross infection from an infectious agent of tissues and cells, removed cells or related materials from removal in clinical application; 3) Hypersensitivity: hypersensitivity reactions, including allergies, anaphylactoid reactions or anaphylaxis; 4) Malignancy: malignant diseases possibly transported by tissues/cells (irrespective of origin, donor or procedure); 5) Rejection: unexpectedly delayed or absent implantation, graft failure (including mechanical damage); 6) Toxicity: toxic effects from tissues and cells or related materials; 7) Incompatibility: unexpected immune reactions due to incompatibility of tissues/cells; 8) Unjustified-abusive-excessive risk: invalid procedure, which includes the unnecessary exposure to risk, e.g. error of tissue provided, which was discovered after the patient’s anesthesia and with the surgical procedure having begun; 9) Genetic abnormality: presumed transmission of genetics disease; 10) Other transmission: presumed transmission of other (non-infectious) diseases.
Other reactions may also be noted (as in the case of HPC transplantation), unexpected or serious graft-versus-host disease (GVHD) and some transfusion-related reactions, such as hemolytic reaction, transfusion-related acute lung injury (TRALI) or transfusion-associated circulatory overload (TACO). Note that the above reactions are indicative, other reactions may also occur.
In conclusion, the tissue establishments procuring tissues and cells should provide to clinical users clear guidelines regarding the way to report adverse reactions, using preferably standardized documentation. In general, the presumed adverse reactions should be directly reported by clinical users to the tissue establishment that provided those tissues or cells before the investigation or confirmation of the reaction. This approach allows the tissue establishment to be involved in research process by assuming the appropriate measures and preventive actions to prevent damage in other patients [13].
Allocation and assessment of adverse reactions
The allocation of SARs is defined as “the probability of a serious adverse reaction at a recipient to be attributed to tissues or cells that are transplanted or of a serious adverse reaction to a living donor to be attributed to the donation process” [18].
The allocation of a SAR can be changed during research, as data are collected. In case of SARs at recipients, the evidence may be related to the inter connection between the recipient condition and one characteristic of the tissues or cells transplanted or identification of a comparable situation in the donor. Alternatively, it could be related to the identification of other possible sources or causes of the condition of the recipient. The scale for reaction allocation, referred to in Table 1 [14] is included in the submission guidelines of annual reports in the EC.
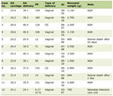 Click to view | Table 1. Allocation Scale of Serious Adverse Reactions (SARs) |
However, the assessment of the allocation of SAR carried out before report submission to the CA should be considered preliminary and should not delay report submission. In particular, it is not advisable for a report being delayed until the results of laboratory tests are obtained. The preliminary assessment is carried out only to avoid reporting those cases, where the allocation is driven by the result of “exclusion” (see allocation scale). Report submission without the assessment is better than a subsequent report [19].
The ranking table referring to Table 2 [14] applies only to the assessment of serious reactions. Because different systems exist, and new models are developed for assessing the severity of a reaction and in connection with the system of the International Society for Blood Transfusion (ISBT), a system was developed for the suspicion of a serious reaction from a transmissible infectious agent (bacterium, fungus, virus, parasites), and was then adopted by the EC. Only SARs at recipients which are graded as “severe”, life-threatening ones or death should be reported in CA (Table 2).
Click to view | Table 2. Evaluation of Serious Adverse Reactions (SARs) |
| Adverse Events Report | ▴Top |
The SAE is defined in European legislation as follows: “A serious adverse event is considered to be every unfortunate incident associated with the procurement, testing, processing, storage and distribution of tissues and cells and which could lead to the transmission of an infectious disease, to death or a life-threatening disability or incapability of patients or could cause or prolong hospitalization or morbidity” [11].
Categories and allocation of adverse events
For reports submitting to EC, SAEs are divided into four categories: 1) Tissue and cell deficiency (this should be understood as an inherent defect at tissues or cells, rather than emerging at procurement, processing, storage, distribution); 2) Equipment damage; 3) Human error; 4) Other (this category includes any kind of procedure failure from procurement to clinical application).
Adverse events can be detected at any stage of the process from donation to transplantation. CAs do not need to be informed for every deviation from the standard operating procedures (SOP) within the tissue establishment. The 2006/86/EC [12] directive clarifies that only “severe” adverse events should be reported to the CA. Events without obvious damage possibility (negligible impact) should be collected and monitored at the hospital or at the tissue establishment level, as they might suggest deficiencies in the quality of service offered. The CA usually does not collect or even locate these events, unless multiple errors have been reported. This may indicate a systematic failure.
The 2004/23/EC [11] directive defines SAEs with regard to the probability of causing SAR. The severity can be associated with the probability of severity of an adverse reaction, if the adverse event had not been discovered, or the severity of the adverse reaction which could occur as a result of an isolated event in another location or time. When a SAE derives from an isolated event but has consequences for multiple products, it should be reported as a SAE.
Deviations from SOP at tissue establishments or other adverse events having consequences on the quality and safety of tissues and cells, should lead to report of SAE to CA, even and if the event took place only at one tissue establishment, when one or more of the following criteria applies: 1) Inappropriate tissues/cells have been distributed for clinical use, even if not used; 2) The event could have consequences for other patients or donors due to common practices, services, procurements or donors; 3) The event has resulted in a confusing complication of tissues or cells; 4) The event has resulted in the loss of irreplaceable autologous tissues or cells or high compatibility (e.g., specific receptors) allogeneic tissues or cells; 5) The event resulted in a significant loss of incompatible-non-compatible allogeneic tissues, or cells. A “significant quantity” should be considered as the quantity that affects the treatment of the patient. Therefore, it will be lower for these tissues or cells at a low offer level and higher for those at abundant-high offer.
Consequently, since the criteria listed above are met, adverse events can be considered as posing a serious risk to patient’s health and under these circumstances should be reported to the CA.
Assessment of adverse events severity
Table 3 [1-5] is a non-exhaustive list of several types of adverse events that could be reported.
Click to view | Table 3. Evaluation of Adverse Events Severity [1-5] |
| The Biovigilance System | ▴Top |
Quality and safety standards implementation to tissues and cells
In recent years, the demand for medicinal products of human origin is steadily increasing with the development of new therapeutic applications, the improvement of access to health care and the change in demographic details of the potential donors/recipients, such as aging and chronic conditions. For this reason, the level of safety, efficiency and quality of MPHO, as exceptional health care products, should be maintained and continuously optimized. This requires the implementation of quality systems, including traceability and biovigilance, both at national and supranational level, due to the continuously increasing transnational exchange of patients and human products [20].
In order for the risks to be mitigated and high safety levels of public health protection to be assured, quality and safety standards are required at all stages of the process, from donation, which will then lead to transfusion or human implementation or transplantation of the MPHO. For this reason, guidelines have been drawn in order to determine common (minimum) quality and safety standards at the EU level, which aim at facilitating increased transnational exchange of said substances, concurrently allowing regulation of clinical application and ethical issues there of (such as donor consent) to be modulated at national level.
However, when MPHO are further used for the manufacture of drugs or medicinal products, the protection of public health is ensured with combined use of directives which are initially implemented at donation, procurement and testing of MPHO implemented as raw materials in combination with the legal framework of drug/medicinal products.
The application of quality and safety standards, the development of medical protocols and cooperation protocols between member states, the implementation of the European code and the development of electronic traceability systems aim at vigilance and in the surveillance of the MPHO from donation up to the transplantation thereof [15].
Biovigilance system development
A prerequisite for combining the above actions is the development of a biovigilance system. Biovigilance includes all organized processes concerning detection, evaluation and monitoring, which are associated with adverse events and reactions observed at donors or recipients, as well as with epidemiological follow-up of donors/recipients.
This system operates at three institutional levels: 1) At the European level, where the EC exerts, inter alia, coordinating and supportive role, maintaining the early warning system for serious adverse reactions and events (SARE) (rapid alert) for tissues and cells, as well as the European Center for Disease Control (ECDC), which monitors all adverse events and threats concerning public health; 2) At the national level, where the CAs assume the difficult role of ensuring that the EU Directive requirements are met, by developing frameworks of quality and safety; 3) At the local level where hospitals, tissue and cell establishments, organizations and primary health care providers should apply all the required quality and safety standards through SOPs [10].
WHO in an attempt to optimize services concerning the MPHOs identified three international governmental approaches, which were developed through long-term cooperations of scientific carriers and CA for improvement and harmonization access to safe, effective and ethical applications at national, regional and local level [1].
These approaches concern: 1) The consensus and implementation of a group of common guidelines for all MPHO; 2) The global use of the International Society for Blood Donation and Transplantation (ISBT128), to enhance traceability and transparency throughout the world; 3) The maximum exchange of information of V&S at global level.
The scope of biovigilance is quite extended and includes donation up to recipient follow-up post MPHOs appliance. The individual application fields of biovigilance concern activities related to testing, processing, storage/maintenance, release, import/export and distribution of MPHO. The risk of disease transmission may be significantly reduced at initial stages of the process, by the collecting/procurement organizations of MPHOs, with careful update and selection of the donor, by obtaining detailed medical history and with the necessary laboratory testing according to the application of procedures as also defined from the relevant EU guidelines for organ [20], tissues and cells [11, 12, 21, 22] and blood [18] procurement with their amendments. Certainly, these quality assurance systems require that the applied methods and laboratory protocols require extensive methodological standardization steps, i.e. detailed determination of sensitivity, specificity, positive and negative predictive values, inter-assay and intra-assay variation, interobserver variability etc [23, 24].
Key parameters of a biovigilance system
Effective MPHO biovigilance systems mainly concern the reports of the clinicians, who are responsible for submitting reports not only for recipient’s SARs (associated with the graft) but also for donor’s SARs, which are not however related to this.
The biovigilance system should be incorporated in the context of quality management program of the CA, with one or more SOPs, which should describe: 1) The recognition process of the reports received; 2) The maintenance of the reports to the CA files; 3) The identification of the SAR to the product applied; 4) The root cause analysis; 5) The assessment of the results of root causes; 6) The rapid alert of other institutions at national or international level; 7) The follow-up of corrective and preventive actions at a later inspection of the CA, or earlier, where necessary; 8) The annual submission report to the EC regarding the reports received.
The biovigilance responsible(s) of the CA should to be trained for said function [19]. Finally, standard operating procedures should describe the rapid action taken by all the organizations concerned about the safety of recipients. This may include tissue and cell quarantine, recall, and look-back or follow-up of the patients where MPHO have already been transplanted.
The nodal points of effective biovigilance systems
Within the framework of the EU funded project EUSTITE (www.eustite.org), which included a consortium of organizations of 10 member states in cooperation with WHO, guided by the National Transplantation Center of Italy, the SAREs issue is being investigated which are reported to the EU and tools and guidelines are developed for supporting member states.
The aim of the EUSTITE project was: 1) Promoting standardization of good practice for inspection of tissue establishments; 2) Development of optimal systems for notification and management of the adverse events and reactions associated with the quality and safety of tissues and cells, which are implemented at patients within the EU, regardless of whether tissues and cells come from within the EU or from the third countries. These particular tools were then incorporated in the EC directives towards member states for the submission of SARE reports on an annual basis.
EU directives for tissues and cells determine certain basic key-types in organization of the procedures that should play critical roles in SAREs notification within a member state. The directives also describe how adverse events and reactions should be reported when they are associated to cells and tissues originating from another member state or being imported into the EU from a third country.
The tissue establishment is the reference point for receiving the reports of adverse events and reactions. The tissue establishment is in charge of supporting the notification of adverse events and reactions, providing detailed information in an appropriate language regarding how to report adverse events or reactions: 1) In POs; 2) In tissues and cells ORHAs; 3) In other relevant tissue establishments or 4) In providers who use tissues or cells to produce advanced therapy medicinal products (ATMP).
The 2006/86/EC [12] directive clarifies that the role of tissue establishment does not exclude a PO or ORHA from also reporting-notifying directly to the CA, if necessary. CA is responsible for the creation of the national (or regional) framework of submitting SAREs reports. The CA should provide tissue establishments with guidelines, forms and instructions regarding submission of SARE reports according to national requirements.
The tissue establishment is responsible for providing clear guidelines, forms and instructions to clinical users, removal organizations, as well as to important third institutions on how to notify SAREs in accordance to national or local requirements. The report and management of a SARE should be integrated in the quality system of the tissue establishment, with one or more SOPs describing the report recognition process, research, follow-up of corrective and preventive actions and report to the CA.
Procedures should include management of SAEs detected within the tissue establishment itself. Procedures should allow rapid action uptake by all concerning organisms to protect the safety of recipients. This may include tissue and cell quarantine, recall and look-back of patients at whom tissues or cells have already transplanted. These actions may require to be undertaken by other organizations, other than the one who received the original notification. Also, a series of actions could be taken in case of reporting of a (presumed) potential transmission from a dead organ and cells donor.
| Management of SAR and SAE | ▴Top |
Reporting and investigating adverse events and reactions: organization responsibilities
European directives for tissues and cells recognize a variety of organizational models that need to play a role in managing adverse events in a member state. Also, the ways of reporting said events when the products originate from another member state or from a third country are described. The tissue/cell establishment is the reference point for receiving reports of adverse events.
The first task of the tissue/cell establishment is informing the collaborating institutions with clear and understandable way on how to report events. The collaborating institutions are POs, ORHAs, other relative tissue/cell establishments or advanced drug manufacturers.
Upon receiving a report, the tissue/cell establishment will investigate it further in order to identify the cause and appreciate the severity in cooperation with the PO and ORHA, will inform the CA as it should and will take specific preventive or corrective measures. The CA should confirm that the appropriate measures have been taken. According to the provisions there should be a mechanism ensuring the circulation of information within the EU via the CA network. Of course, the relevant provision does not prohibit any PO or ORHA to update the CA directly [13].
Reporting and investigation of adverse events and reactions per institution are listed in Table 4.
Click to view | Table 4. Reporting and Investigation of Adverse Events and Reactions per Institution |
Alarm-rapid alert
Different alarm models and information mechanisms may be used by the CA for ensuring the quality of tissues and cells and in order to inform others involved in the process. These reactions may be a response to specific SAE/SAR or due to information obtained from various sources, such as other CA, EC, etc.
Rapid alerts are immediate, direct and urgent notifications from or through the CA in a member state to inform other organizations of possible risk. They should be issued only in exceptional cases. At national level they are coordinated by the CA, while at community or international level are issued in cooperation with another CA, the European Commission or WHO.
The following criteria should be met for issuing rapid alert in every member state: 1) SAE/SAR, of serious or likely severe nature; 2) Possible risk to other persons in another member state; 3) Wider public health implications. A contact list has been formulated by the Commission at each CA with people responsible for receiving quick reports and is used when there is an alarm.
| Conclusions | ▴Top |
In recent years, the demand for medical products of human origin is steadily increasing with the development of new therapeutic applications and change in demographic details of potential donors/recipients, such as aging and chronic conditions. However, residual risks or errors result in failures, transmissible diseases or conditions in which donors or patients/recipients are exposed to risks, even if they have not been damaged. Therefore, it is self-evident that the level of safety, efficacy and quality of medical products of human origin, as exceptional healthcare products, must be ensured and continuously optimized with implementation of quality systems, including traceability and biovigilance in both national and supranational level.
In this context, EU member states should ensure, taking appropriate measures, the development of the biovigilance system for reporting, recording and transmission of relevant information regarding SARE that may affect quality and safety of tissues and cells and which are likely attributed to procurement, testing, processing, storage and distribution of tissues and cells, as well as any other SAR found during or after clinical application, which is may be associated with the quality and safety of tissues and cells. The biovigilance system should be integrated in the context of the quality administration project of the competent institutions and in particular of the national CA, with one or more standards of standardized operating procedures.
In order to mitigate risks and ensure high-levels of public health protection, quality and safety standards are required at all stages of the process, from donation, which will then result in transfusion or human implementation or transplantation of medical products of human origin. By adopting the directives of EU common (minimum) quality and safety standards have been established, which aim at facilitating the increased transnational exchange thereof, allowing at the same time regulation of the clinical application thereof and ethical issues (such as donor consent) to be regulated at national level.
The EU directives for tissues and cells recognize various organizational models that exert a key role in managing adverse events in a member state. In these the ways of reporting SARE are also described, when products come from another member state or a third country. Central reference point for receiving the relevant reports consists the tissue/cell establishment. Alarm models and information mechanisms are used by the CA for ensuring quality of tissues and cells. For immediate and urgent updates rapid alerts are issued to exceptional cases in a member state for informing other organisms about potential risk. At national level they are coordinated by the CA, while at community or global level they are issued in cooperation with another CA, the EC or WHO.
The II, PO and ORHA, in cooperation with the CA, should promote a reporting and notifying profile of SAREs, without been related to guilt or punishment but, on the contrary, providing the ability of learning and improvement. A nodal point for successful development of the biovigilance system consists, as a necessary and sufficient condition, the realization of health care professionals of the potential consequences that can cause adverse reactions and adverse events in health and safety of the donor and the recipient. Clinicians are encouraged to be alert of the potential clinical conditions that could be caused by the cells and tissues, monitoring with particular attention the adverse reactions.
Conflict of Interest
MN has received grants by the Deutsche Forschungsgemeinschaft (DFG) through the Sonderforschungsbereich Transregio 19 “Inflammatory Cardiomyopathy” (SFB TR19) (TP B2), and by the University Hospital Giessen and Marburg Foundation Grant “T cell functionality” (UKGM 10/2009). MN has been consultant to the IKDT (Institute for Cardiac Diagnosis and Therapy GmbH, Berlin) 2004 - 2008, and has received honoraria for presentations and/or participated in advisory boards from Abiomed, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Fresenius, Miltenyi Biotech, Novartis, Pfizer and Zoll. All other authors declare no potential conflict of interest.
Abbreviations
ATMP: advanced therapy medicinal products; CA: competent authority; EC: European Commission; ECDC: European Center for Disease Control; EU: European Union; GVHD: graft-versus-host disease; ISBT: International Society for Blood Transfusion; MARU: Medically Assisted Reproduction Units; MPHO: medical products of human origin; ORHA: organizations responsible for human application; QMS: quality management system; SAE: serious adverse event; SAR: serious adverse reaction; V&S: vigilance and surveillance; WHO: World Health Organization
| References | ▴Top |
- Italian National Transplant Center. Vigilance and surveillance for human cells, tissues and organs. The notify booklet vigilance and surveillance (V&S) of medical products of human origin (MPHO). Notify Library the Global Vigilance and Surveillance Database for Medical Products of Human Origin, Geneva, 2013.
- Noel L, Martin DE. The exception of medical products of human origin: Towards global governance tools. In: Rainhorn JD, El Boudamoussi S (eds). New cannibal markets: Globalization and commodification of the human body. Edition Foundations Maison des Sciences de l'Homme, Paris. 2015:383.
doi - EUR-Lex. Report of the European Economic and Social Commission for the "Directive proposal of the European Parliament and of the Council regarding establishing quality and safety standards for donation, procurement, testing, processing, storage and distribution of human tissues and cells". COM(2002)319 final - 2002/0128 (COD) 2003/C85/14.
- World Health Organization Secretariat. Principles for global consensus on the donation and management of blood, blood components and medical products of human origin. WHO Secretariat, Geneva, EB140/18/29.12.2016.
- European Union Legislation on Blood, Tissues, and Cells. Evaluation and fitness check (FC) roadmap. Available at: http://ec.europa.eu/smart-regulation/roadmaps/docs/plan_2016_154_evaluation_eu_legislation_on_blood_en.pdf.
- Oliva MS, Schottman T, Gulati M. Turning the tide of corneal blindness. Indian J Ophthalmol. 2012;60(5):423-427.
doi pubmed - Lakey JR, Mirbolooki M, Rogers C, Mohr J. Supply of human allograft tissue in Canada. Cell Tissue Bank. 2007;8(2):135-150.
doi pubmed - Ping P, Zhu WB, Zhang XZ, Li YS, Wang QX, Cao XR, Liu Y, et al. Sperm donation and its application in China: a 7-year multicenter retrospective study. Asian J Androl. 2011;13(4):644-648.
doi pubmed - Martin D, Kane S. National self-sufficiency in reproductive resources:An innovative response to transnational reproductive travel. Int J Fem Approaches. 2014;7:10-44.
- World Health Organization. Notify report - Part A: Exploring vigilance notification for organs, tissues and cells. A global consultation organized by CNT with the co-sponsorship of WHO and the participation of the EU-funded SOHO V&S project. Bologna. 2011.
- Directive 2004/23/EC. European Parliament and Council Directive of the 31st of March 2004 for establishing types of quality and safety for donation, procurement, testing, processing, maintenance, storage, and distribution of human tissues and cells. Official Journal of the European Union L102/48, 7.4.2004.
- Directive 2006/86/EC. Commission Directive of the 24th of October2006 for the application of 2004/23/EC directive of the European Parliament and Council with regard to traceability requirements, notifying serious adverse reactions and events, as well as certain technical requirements for coding, processing, maintenance, storage and distribution of human tissues and cells. Official Journal of the European Union European L294/32, 25.10.2006.
- European Directorate for the Quality of Medicines & Health Care of the Council of Europe. Guide to the quality and safety of tissues and cells for human application. 3rd. EDQM. 2017.
- Assisted Reproductive Technologies and Haematopoieticstem Cells Improvements for Quality and Safety Throughout Europe. Deliverable 8: Guidance for establishing a hematopoietic progenitor cells donor follow-up registry (workpackage 5). This project is co-funded by the EU Public Health Programme; grant agreement no 20132101. ARTHIQS. 2013.
- Italian National Transplant Centre. Survey of European Vigilance& Surveillance Systems (work package 4). This project is supported by the EU Public Health Programme; grant agreement no 200091110. SOHO V&S, 2011. Available at: http://www.notifylibrary.org/background-documents#VIGILANCEANDSURVEILLANCEREPORTS.
- Alvarez Μ, Garrido G, Fehily D, Delvecchio C, Nanni Costa A, Matesanz R. Tissue and cell inspection systems in Europe: AEUSTITE survey. Organs, Tissue and Cells. 2008;2:87-89.
- Directive 2005/61/EC. Commission Directive of the 30th of September 2005, for application of 2002/98/EC directive of the European Parliament and Council regarding traceability (detectability) requirements and reporting of serious adverse reactions and events (EEL 256/32, 1.10.2005). Available at: http://eurlex.europa.eu/legal-content/EL/TXT/ ?uri=CELEX:32005L0061.
- SOHO V&S PROJECT. Deliverable 8: Communication and investigation of serious adverse events and reactions associated with human tissues and cells. Vigilance and Surveillance of Substances of Human Origin (SOHO V&S). A project funded by the EU Public Health Programme (project no: 20091110). SOHO V&S. 2013.
- Directive 2012/25/EU. Commission Directive of the 9th of October 2012, for establishing procedures for information regarding exchange between Member States of human organs intended for transplantation (EEL 275/27, 10.10.2012). Available at: http://eur-lex.europa.eu/legal-content/EN/TXT/ies=CELEX: 52016DC0809.
- Directive 2015/565/EU. Commission Directive of the 8th of April 2015, for amendment of 2006/86/EC directive regarding certain technical requirements for coding human tissues and cells (EEL 93/43, 9.4.2015). Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=%20uriserv%3AOJ.L_.2015.093.01.0043.01.ENG.
- Directive 2015/566/EU. Commission Directive of the 8th of April2015, regarding the application of 2004/23/EC directive concerning procedures for verifying equivalence the quality and safety standards of imported tissues and cells (EEL 93/56, 9.4.2015). Available at: https://eur-lex.europa.eu/legal-content/EL/%20TXT/?uri=CELEX%3A32015L0566.
- Directive 2006/17/EC. Commission Directive of 8th of February2006, regarding the application of 2004/23/EC directive of the European Parliament and Council concerning certain technical requirements for donation, procurement and testing of human tissues and cells. Official Journal of the European Union (L38/40, 9.2.2006). Available at: http://eur-lex.europa.eu/legal-content/EL/TXT/?uri=CELEX%3A32012L0039.
- Noutsias M, Pauschinger M, Ostermann K, Escher F, Blohm JH, Schultheiss H, Kuhl U. Digital image analysis system for the quantification of infiltrates and cell adhesion molecules in inflammatory cardiomyopathy. Med Sci Monit. 2002;8(5):MT59-71.
pubmed - Noutsias M, Rohde M, Block A, Klippert K, Lettau O, Blunert K, Hummel M, et al. Preamplification techniques for real-time RT-PCR analyses of endomyocardial biopsies. BMC Mol Biol. 2008;9:3.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.