

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 9, Number 9, September 2017, pages 812-819
Importance of Distinguishing Between Mitochondrial Encephalomyopathy With Elderly Onset of Stroke-Like Episodes and Cerebral Infarction
Syuichi Tetsukaa, c, Asako Tagawab, Tomoko Ogawab, Mieko Otsukab, Ritsuo Hashimotob, Hiroyuki Katob
aDepartment of Neurology, Hospital of Yuki, 9629-1, Yuki, Yuki-City, Ibaraki 307-0001, Japan
bDepartment of Neurology, Hospital of International University of Health and Welfare, 537-3, Iguchi, Nasushiobara, Tochigi 329-2763, Japan
cCorresponding Author: Syuichi Tetsuka, Department of Neurology, Hospital of Yuki, 9629-1, Yuki, Yuki-City, Ibaraki 307-0001, Japan
Manuscript submitted June 29, 2017, accepted July 21, 2017
Short title: MELAS as Differential Diagnosis of Infarction
doi: https://doi.org/10.14740/jocmr3122w
| Abstract | ▴Top |
The most common disease-causing mitochondrial DNA (mtDNA) mutation in mitochondrial encephalomyopathy (ME) with lactic acidosis and stroke-like episodes (MELAS) is m.3243A>G. In the future, the incidence of patients with cerebral infarction and diabetes mellitus is expected to increase tremendously. Additionally, the A3243G mutation typical of diabetes is estimated to be present in approximately 2% of all diabetes patients, which suggests that the potential disease population with a mitochondrial disorder is greater than previously thought, and there may have been many cases among the elderly that were misdiagnosed. Considering this background, MELAS with the onset of stroke-like episodes should be considered an important differential diagnosis for elderly patients with cerebral infarction, although it might have been overlooked until now. A 68-year-old Japanese female developed convulsive seizures and was admitted to Hospital of International University of Health and Welfare for epilepsy. She had been hospitalized twice in the previous year for cerebral infarction and seizures. She experienced sensorineural hearing loss at a young age. Thus, although she was elderly, we suspected MELAS and detected elevations of pyruvic and lactic acid. A genetic test revealed a point mutation in the mtDNA (m.3243A>G) that led to a definitive diagnosis of MELAS. To date, MELAS has been regarded as a disease of the relatively young. The incidence of patients with cerebral infarction and diabetes mellitus is expected to greatly increase. Thus, we should evaluate cerebral infarction in the elderly with caution to prevent missed diagnoses of MELAS.
Keywords: MELAS; Elderly onset; m.3243A>G; Cerebral infarction; Diabetes mellitus
| Introduction | ▴Top |
Mitochondrial disorders consist of a heterogeneous group of disorders that are linked to mutations in mitochondrial DNA (mtDNA) or nuclear mitochondrial maintenance genes. The most common disease-causing mtDNA mutation in mitochondrial encephalomyopathy (ME) with lactic acidosis and stroke-like episodes (MELAS) syndrome is m.3243A>G, which is estimated to account for 80% of mitochondrial disorders [1]. Currently, the definitive diagnosis of MELAS is generally achieved by identifying the m.3243A>G mutation rather than through the performance of muscle biopsies. This mutation has been reported to occur in approximately 2% of adult diabetes mellitus (DM) patients [2, 3]. DM and age are typical risk factors for cerebral infarction. Therefore, there is a high risk of misdiagnosis, which is defined as the failure to correctly recognize MELAS with an elderly onset of stroke-like episodes in cases of elderly cerebral infarction. According to the diagnostic criteria, stroke-like episodes have to appear before the age of 40 in MELAS [4]. Conventionally, “elderly” has been defined as a chronological age of 65 or older, but few cases occur in the elderly above the age of 65 [5, 6]. Japan is currently an aging society with a large proportion of elderly citizens. Consequently, the incidence of cerebral infarction is predicted to increase in the future. Therefore, MELAS with the onset of stroke-like episodes should be considered an important differential diagnosis for elderly patients with cerebral infarction, although it might have been overlooked until now.
| Case Report | ▴Top |
A 68-year-old Japanese female developed convulsive seizures after sudden sweating during the early morning of April 10, 2016. The convulsive seizures subsided after approximately 3 min. Approximately 1 week before the convulsive seizures occurred, the patient experienced appetite loss and registered a fever with a temperature of 38 °C on April 9. A visit to her doctor led to the suspicion of a urinary tract infection (UTI) for which the patient was prescribed antibiotics. The patient was initially admitted to Hospital of International University of Health and Welfare for suspected pneumonia, UTI, and epilepsy caused by cerebral infarction. A physical examination revealed a height of 153 cm, a body weight of 33.4 kg, a body mass index of 14.3 kg/m2, a body temperature of 37.9 °C, a heart rate of 124 bpm, a blood pressure of 106/89 mm Hg, an SpO2 in room air of approximately 85%, and a Glasgow coma scale score of E3V3M4. The patient occasionally opened her eyes but was unresponsive. She had already recovered from her seizures, which had abated by the time she arrived at our hospital. A chest examination revealed no rattling sound in the lungs, an abdomen examination revealed no reddening at the site of a percutaneous endoscopic gastrostomy, and limb examinations revealed flexion contracture of the left upper limb. The muscle strength was difficult to evaluate, and behavioral observation indicated a manual muscle testing grade of 3-4 in the upper limbs. The grip strength was also difficult to examine. However, the patient was capable of gripping a plastic bottle.
The muscle tone was enhanced on both sides and to a greater extent on the left than the right. Her activities of daily living (ADL) were registered as follows: higher function, capable of communicating through simple conversation, required assistance setting and tidying up for meals, capable of feeding herself using a spoon and fork, required full assistance with transfer to and from a wheelchair, required full assistance with grooming, capable of wiping her face if given a towel, used a portable toilet but required full assistance, used a wheelchair to move about, and required full assistance with dressing, defecation, and urination.
The patient previously worked as a nurse at a hospital between the ages of 18 and 25 years. At age 25, she married and quit her job to become a full-time housewife. She gave birth to a daughter at age 26 (her daughter’s current age is 43) and a son at age 28 (her son’s current age is 41). Her children are both unmarried and have no children of their own. At age 41, she began treatment for diabetes. At age 50, she began using a hearing aid. At age 67 (in May 2014), she was admitted to another hospital for a cerebral infarction (left temporal lobe; Fig. 1a, b). No evident paralyses of the limbs were noted, but the patient did exhibit a disturbance of consciousness. After admission, the patient experienced convulsive seizures. In October 2014, the patient was admitted to the same hospital for the same symptoms and received treatment for a cerebral infarction (right temporal lobe; Fig. 2a, b). In June 2015, the patient was discharged, and since that time, her impairment in terms of regarding ADL and her cognitive dysfunction have progressed rapidly. The patient’s mother had a loss of hearing, used a hearing aid, and lived to 96 years of age. The patient has seven siblings and is the fourth-born child. Her third-born sister died of kidney failure at age 19. Her other siblings are still alive, and the eldest sister is 83 years of age and is independent in terms of ADL.
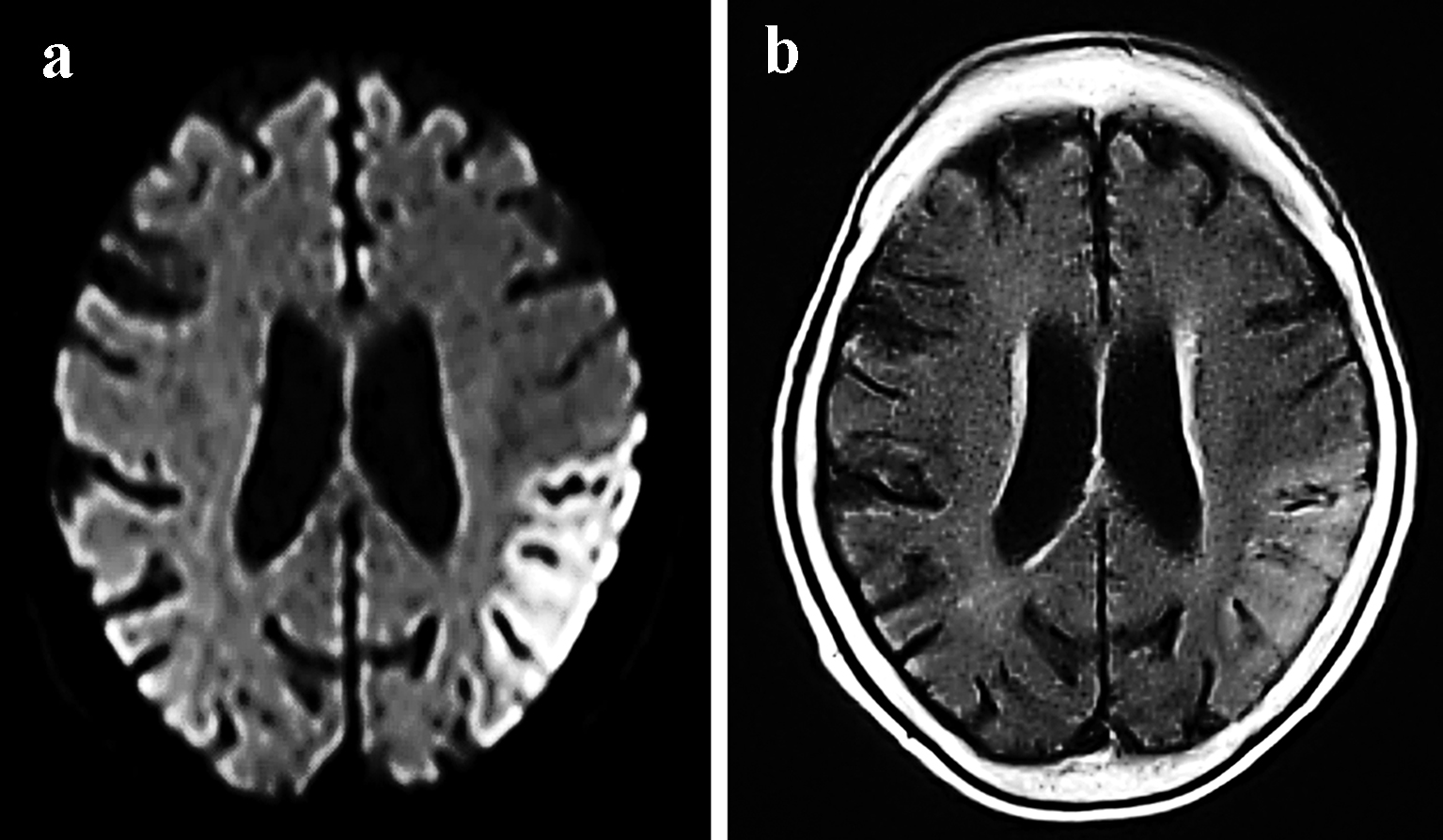 Click for large image | Figure 1. MRI examinations from May 2014 showing the migratory lesions of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Cortical DWI (a) and FLAIR (b) sequences revealed hyperintensity in the left temporal lobe. |
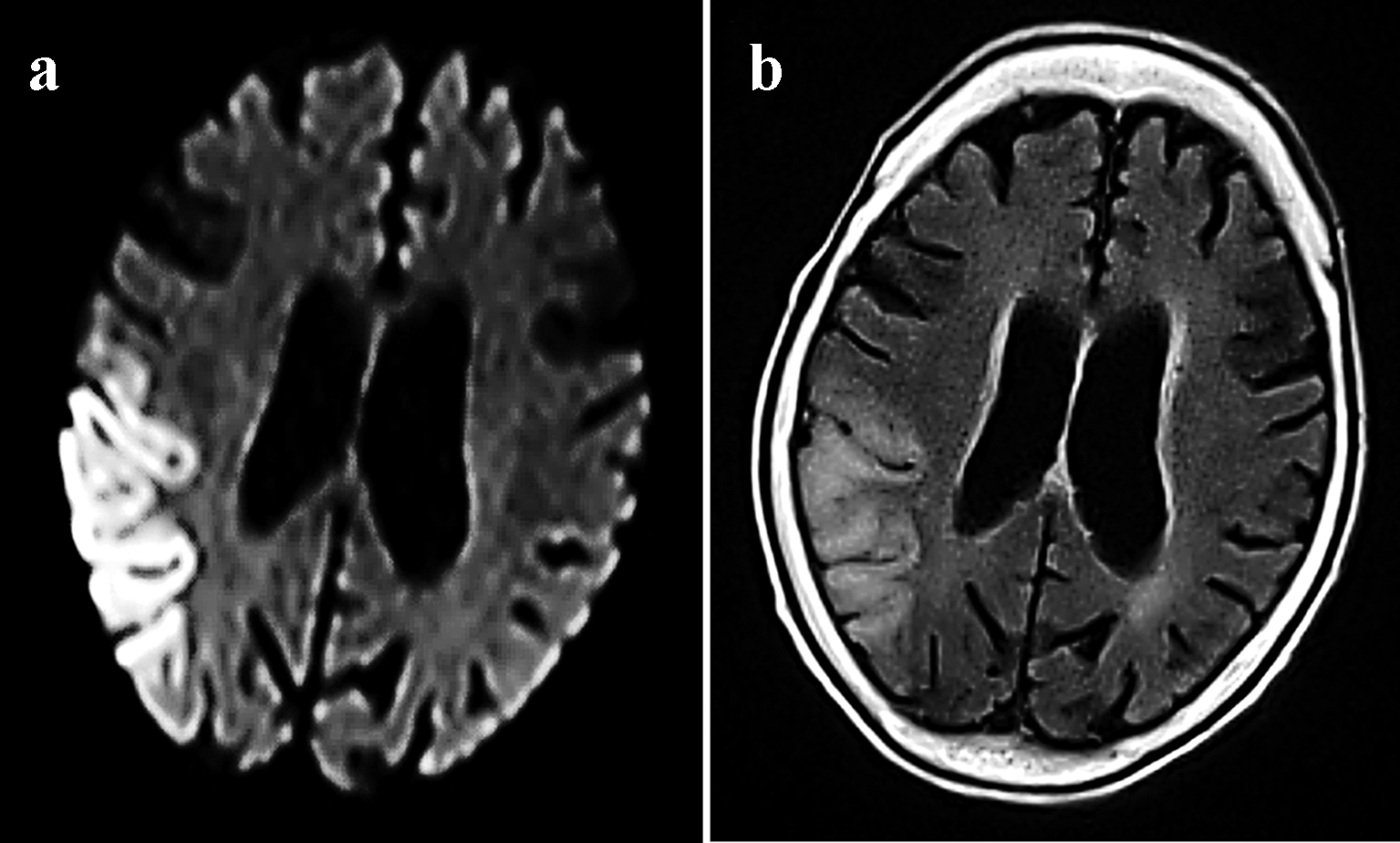 Click for large image | Figure 2. MRI examinations from October 2014 showing the migratory lesions of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Cortical DWI (a) and FLAIR (b) sequences revealed hyperintensity in the right temporal lobe. |
After admission to our neurology department, the patient underwent fluid replacement and was started on ceftriaxone for infection and fosphenytoin sodium hydrate intravenous injections for convulsive seizures for a week. Thereafter, oral phenytoin was titrated to 200 mg/day. A computed tomography (CT) scan of the head taken on April 2016 revealed calcification around the basal ganglia (Fig. 3a, b). The patient had originally been initiated on insulin at our hospital for diabetes, and a loss of hearing was noted at this time. Echocardiography did not reveal any significant disorders outside of myocardial disease. Magnetic resonance imaging (MRI) scans of the head that were performed during all hospitalizations also revealed cortical laminar necrosis-like findings, white matter and the cortical atrophy, which was recognized prominently in the cerebellar cortex (Fig. 4a, b). Hypocalcemia was observed on blood tests, which, in addition to the patient’s history of convulsive seizures, suggested parathyroidism and late-onset MELAS. A blood test revealed an intact parathyroid hormone level of 29 pg/mL that was within the normal range, but the test also revealed elevated pyruvic acid (1.51 mg/dL) and lactic acid (32.9 mg/dL) levels, which strengthened the suspicion of MELAS. To make the diagnosis of MELAS, we outsourced the genetic testing of her blood. The genetic analysis identified an adenine-to-guanine mtDNA point mutation at nucleotide 3243 (m.3243A>G), which led to a definitive diagnosis of MELAS. The patient’s inflammatory reaction subsequently subsided, and her condition stabilized without the development sudden recurrence or worsening of the seizures in the follow-up period, and the phenytoin was continued.
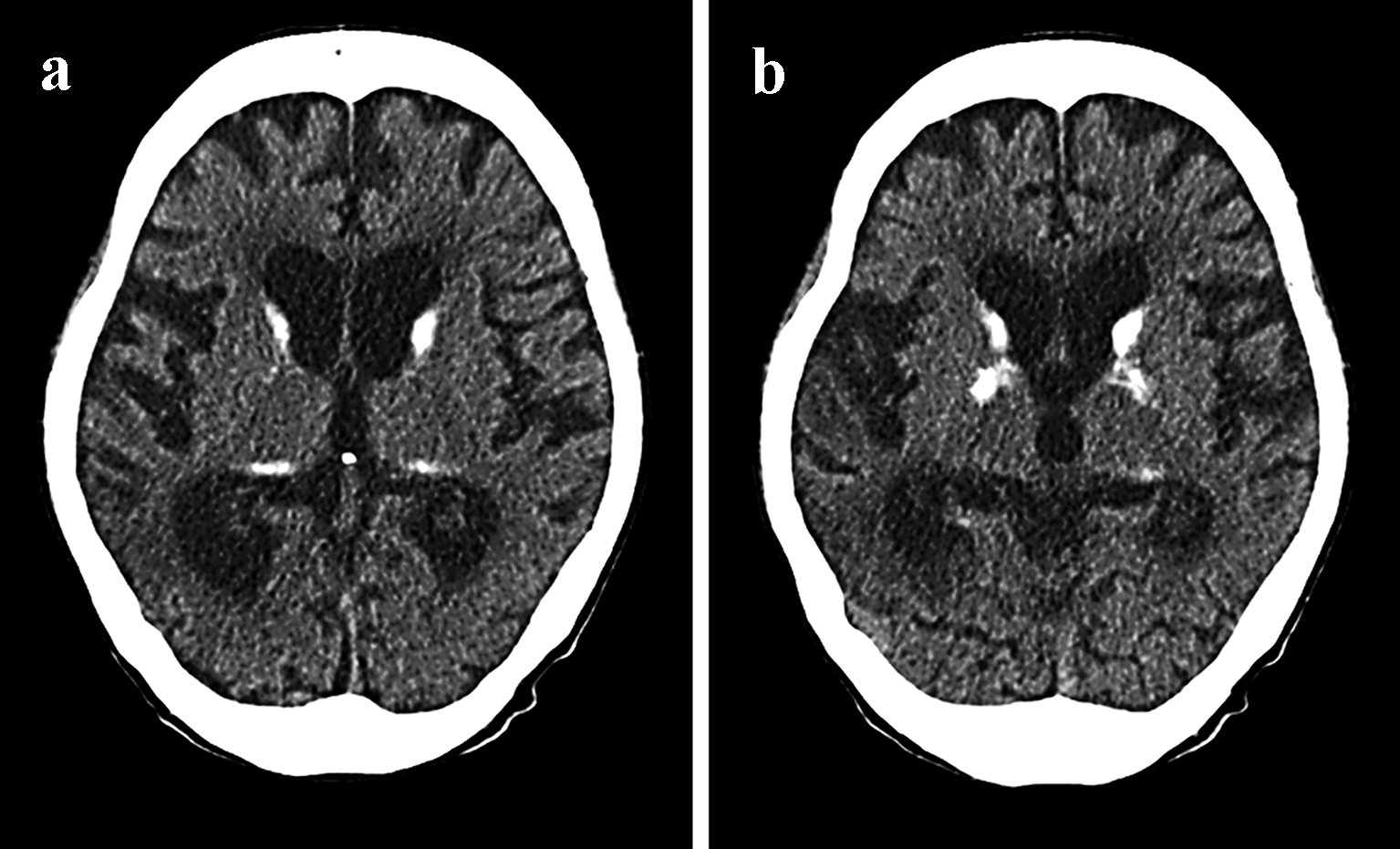 Click for large image | Figure 3. CT scan of the brain from April 2016 of a case with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS). In retrospect, the additional clinical features and bilateral basal ganglia calcification (a and b) were suggestive of MELAS. |
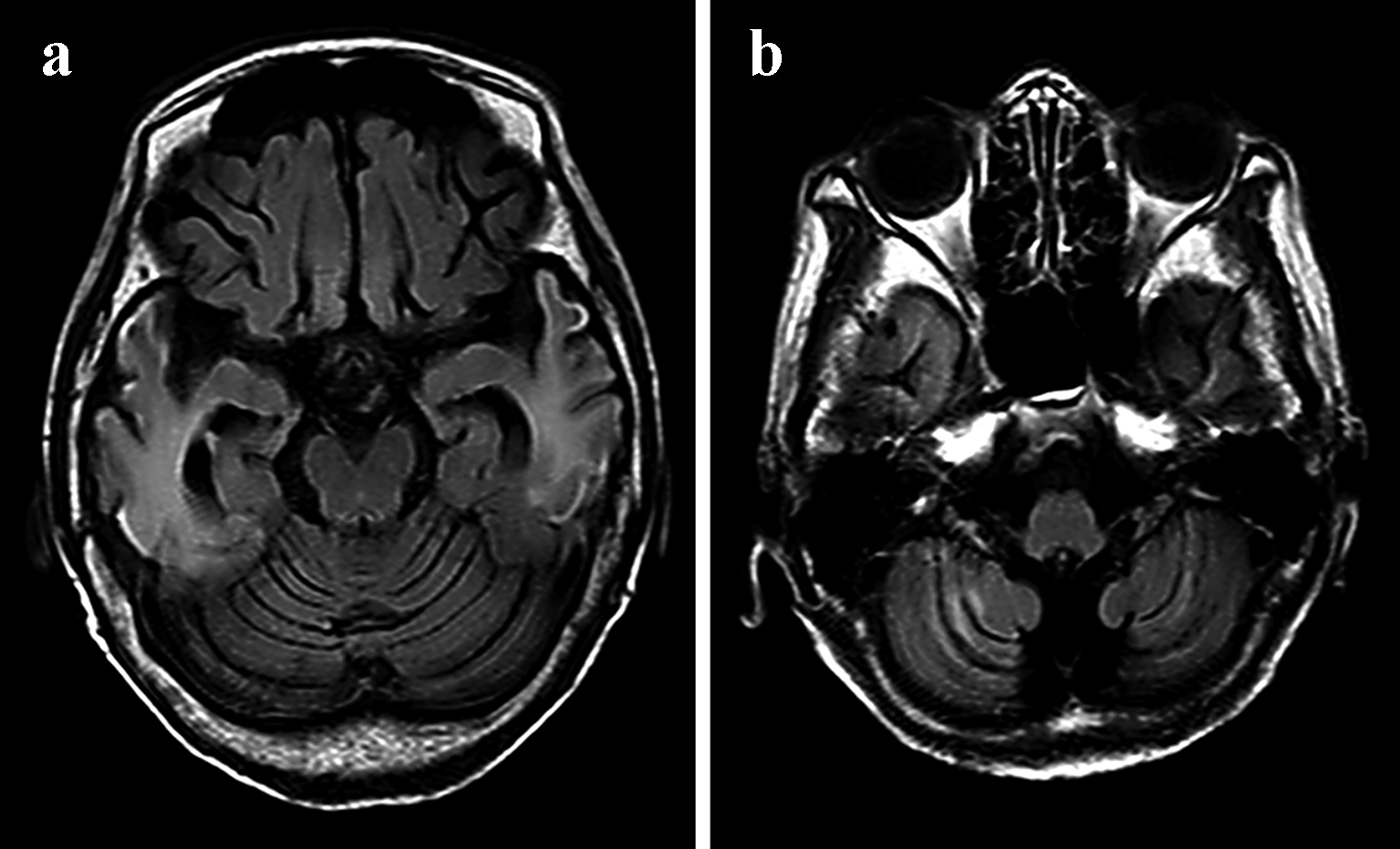 Click for large image | Figure 4. MRI of the brain from April 2016 of a case with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS). FLAIR (a) sequences revealed white matter and cortical laminar necrosis-like findings and the cortical atrophy in the right temporal lobe. (b) The cortical atrophy was recognized prominently in the cerebellar cortex. |
| Discussion | ▴Top |
ME
ME is a generic name for syndromes that exhibit various neurological, muscular, and systemic organ symptoms due to a dysfunction of the mitochondria, which are responsible for energy metabolism in humans. Because ME has a wide range of causes that include dysfunctions of the electron transport chain, enzymes, pyruvate metabolism, tricarboxylic acid cycle-related metabolism, fatty acid metabolism, nucleic acid metabolism, and adenosine triphosphate transfer, it exhibits a diverse range of symptoms in addition to the neurological and muscular symptoms that include cardiac, circulatory, renal, endocrine, gastrointestinal, sensory, and blood symptoms.
MELAS
Before age 40, MELAS is characterized by stroke-like episodes that occur with symptoms that include headache, vomiting, seizures, visual field abnormalities, motor paralysis of the four limbs, and disturbances of consciousness. Head scans (CT and MRI) in the acute phase reveal abnormal findings that resemble those of cerebral infarction in appearance, but the locations of these findings are inconsistent with the main blood vessel-dominant regions of the cerebral artery, and these abnormal regions expand and disappear over the course of the disease as observed in our case. In the early stages of the disease, the above-mentioned symptoms that accompany stroke-like episodes are reversible, but as the seizures recur, these symptoms remain as obvious aftereffects and ultimately result in the atrophy of the infarction-like areas of the brain. The associated complications often include migraine, muscle weakness, emaciation, sensorineural hearing loss, exotropia, drooping eyelids, neuropathy, hypertrophic cardiomyopathy, intraventricular conduction disturbances such as Wolff-Parkinson-White syndrome and De Toni-Fanconi syndrome, diabetes, short stature, and multiple endocrine disorders such as hypothyroidism. The A3243G mtDNA mutation is also observed in 80% of patients. Temporally and spatially, the recurrent stroke-like episodes ultimately result in the patient becoming bedridden, which is similar to the outcomes of recurrent recusant cerebral infarction and cerebrovascular dementia, or result in death from multiple organ failure. Cohort studies of MELAS in Japan have placed the mean ages of death from MELAS at 15 years and 2 months in pediatric cases and 40 years in adult cases [7, 8]. mtDNA disorders with the A3243G mutation typical of diabetes are estimated to be present in approximately 2% of all diabetes patients, which suggests that the potential disease population with mitochondrial disorders is greater than previously thought.
MELAS primarily occurs in childhood, which means that 80% of patients exhibit their first symptoms of seizure between the ages of 5 and 15. The stroke-like symptoms with high incidences are paroxysmal headache and vomiting (in nearly all cases), seizures (≥ 90%), transient hemiplegia, and visual impairment (semi-blindness). During episodes, patients exhibit marked acidosis (serum and spinal fluid lactic acid levels at least 2 - 3 times the normal level), and CT and MRI scans reveal local ischemic changes that primarily occur in the occipital region. Unlike cerebral infarction observed in adults, MELAS presents with low-density areas that are inconsistent with the main blood-vessel-dominant region. However, there is no doubt that some types of vascular lesion are strongly involved in the onset of this disease. Prior to the appearance of seizure-related symptoms, most patients are asymptomatic, and many exhibit no intellectual abnormalities. Nonetheless, short stature, easy fatigability, and mild muscle weakness are often observed. As seizure-related symptoms recur, patients exhibit symptoms that include intellectual deterioration, emaciation, and renal failure, which ultimately lead to death. Research has revealed that 80% of patients with this disease have the 3243 A→G point mutation (3243 mutation) in the region that codes for tRNALeu (UUR) in the mtDNA, and 10% of patients have the 3271 T→C point mutation of the same tRNA [9, 10]. Even if the mother and siblings of a typical MELAS patient carry the same 3243 mutation, the time required for the patient to transition from being asymptomatic to exhibiting typical MELAS symptoms varies [2]. Cases of familial diabetes with the 3243 mutation have also been discovered, and revelations about the various symptoms that the 3243 mutation exhibits are continuing to be made.
The diversity of symptoms is the result of tissue specificity
The coexistence of mutated and normal mtDNAs within a single tissue is a characteristic finding of ME. Moreover, the amount of mutated mtDNA differs both between tissues and between cells (e.g., between muscle fibers in muscle tissue). Muscle fibers that accumulate mitochondria take an abnormal form that is referred to as “ragged red fiber (RRF)”, and the existence of RRF is a diagnostic finding for mitochondrial disease. RRF contains substantial amounts of mutated mtDNA and decreased amounts of normal mtDNA [11]. With the MELAS-associated 3243 mutation, the increase in mutated mtDNA within the vascular smooth muscle cells is a noteworthy finding. The quantity of mitochondria also increases in these abnormal blood vessels. Thus, MELAS involves systemic abnormalities of the vascular system, and these abnormalities are thought to manifest in the central nervous system and trigger stroke-like symptoms. Thus, the performance of a muscle biopsy is ideal for reaching a definitive diagnosis based on the detection of RRF. However, muscle biopsies are invasive examinations that place a burden on the patient. Furthermore, research results related to MELAS and the m.3243A>G mutation are currently accumulating. Thus, if there are sufficient clinical features and accompanying neuroimaging findings, as in our case, it has recently become unnecessary to perform a muscle biopsy.
The prognosis cannot be estimated from the amount of mutated mtDNA
Research in culture systems has revealed that when the mutated mtDNA within a single cell increases beyond a certain threshold, the tissue containing that cell loses function. This threshold is thought to be 90-95% with the 3243 mutation (normal mtDNA at levels of 5-10% is able to compensate for this mutation) [12]. However, no correlation exists between mutated mtDNA and clinical symptoms in terms of the clinically measurable DNA isolated from muscle and blood [10]. This lack of a correlation means that even if a mutation in the mtDNA is present, the future onset of MELAS and the specifics of the associated symptoms cannot be predicted. Furthermore, even if a female carries mutated mtDNA, her children are not 100% certain to also carry that mutation. The difficulty of prenatal diagnoses and genetic counseling for ME is thus becoming increasingly clear.
MELAS is an important differential diagnosis for cerebral infarction with the elderly onset of stroke-like episodes
In the future, it is expected that the incidence of patients with cerebral infarction and DM will increase tremendously. Additionally, the A3243G mutation typical of diabetes is estimated to be present in approximately 2% of all diabetes patients, which suggests that the potential disease population with a mitochondrial disorder is greater than previously thought, and there may have been many previous elderly cases that have been misdiagnosed. Temporally and spatially, these recurrent stroke-like episodes ultimately result in the patient becoming bedridden as occurs due to recurrent recusant cerebral infarction and cerebrovascular dementia. Currently, before the age of 40 years, MELAS is characterized by stroke-like episodes that occur with various symptoms. Thus far, MELAS has rarely been reported to present with stroke-like episodes after the age of 40 years. Additionally, it is uncertain why the onset of neurological dysfunction is delayed into adulthood in some MELAS patients. The pathological mechanism may be due to a lower mitochondrial mutation load in the brains of these patients because the diversity of symptoms is the result of tissue specificity as mentioned above. Specifically, in cases of elderly onset stroke-like episodes such as our case, it is possible that the patients’ brains carry an even lower mitochondrial mutation load. Although there are some published cases of adult-onset patients who presented with MELAS with the m.3243A>G mutation [10, 13-15], elderly MELAS patients over the age of 65 are not commonly reported [5, 6] (Table 1). To the best of our knowledge, the present case is only the third such report. Based on the presently available research results, when a mutation in the mtDNA exists, the onset of MELAS and the specifics of the associated symptoms cannot be predicted.
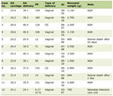 Click to view | Table 1. Summary of Published Cases of Patients Older Than 65 Years Who Presented With MELAS |
The symptoms (i.e., seizures, visual field abnormalities, motor paralysis of the limbs, disturbances of consciousness, muscle weakness, emaciation, sensorineural hearing loss, and progressive higher brain dysfunction) and head scans (CT and MRI) of MELAS patients with stroke-like symptom are very similar to those of cerebral infarction in the elderly. Regarding head scans (CT and MRI), unlike cerebral infarction observed in adults, it is thought that MELAS presents with low-density areas that are inconsistent with the main blood-vessel-dominant region. Additionally, the CT scan that revealed calcification around the basal ganglia and the brain CT that revealed calcification in 2016 provided us with clues regarding the diagnosis of MELAS in practice, although this CT finding is also recognized in a large proportion of the elderly. Although basal ganglia calcification is a common incidental finding upon neuroimaging of the elderly, this sign should lead clinicians to consider an underlying mitochondrial disorder in the presence of other appropriate clinical features [16]. Generally, MRI is thought to show hyperintensity on diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) with a predilection for the parietal, temporal, and occipital cortices without characteristically conforming to a single vascular territory [17]. Points that may exhibit reversible change without evident decrease in the apparent diffusion coefficient (ADC) of acute stage lesions differ from a cerebral infarction. The ADC values of the lesions are reported as a mild increase, unchanged, or mild decrease. Although these differences are presumed to reflect the timing of measurement and differences in the state of the disease, in the early stage of the disease it is at least presumed that angioedema forms the core or coexists as legions. We speculate that it is a special edematous change with a strong effect on the cerebral cortex, reflecting hyperpermeability of capillary blood vessels. Such patterns represent a hallmark finding of MELAS in addition to brain CT scans that reveal calcification. Atrophy of the cerebrum is a common feature of mitochondrial in childhood and adulthood [18]. While tissue atrophy may be slowly progressive in some cases, others that demonstrate rapid progression of atrophy that parallels clinical decline [19]. The neuroimaging feature was recognized in our case (Fig. 4a, b). However, it is difficult to accurately identify MELAS based only on this image finding in elderly patients as observed in our case. Such as seizures, one of the characteristics of the MELAS, are also complications of cerebral infarctions and symptomatic epilepsy. There are some differences between MELAS episodes and “regular” cerebral infarctions episodes which are the sudden loss of sensation on one side of the body, altered sensations on one side of the body, difficulty speaking or understanding speech, sudden weakness or paralysis on one side of the body, etc. Thus, based on the results of previous studies and the results of our case, in cerebral infarction in the elderly, we propose that the features presented in Table 2 should be considered in the differential diagnosis of MELAS. When cerebral infarctions are encountered in patients with these characteristics, the pyruvic acid and lactic acid levels should be measured. That the elderly patients with strokes should be suspected of MELAS is true not in the routine cases of an elderly patient with diabetes, but in cases where there are additional features of a mitochondrial disease, such as seizures which are a common complication of cerebral infarctions, early-onset sensorineural hearing loss, image that are inconsistent with the main blood-vessel-dominant region, etc (Table 2).
Click to view | Table 2. Features to be Considered in the Differential Diagnosis of MELAS in Elderly Cerebral Infarction Patients |
Conclusion
To date, MELAS has been regarded as a disease of the relatively young. However, A3243G mutations typical of diabetes are estimated to be present in approximately 2% of all diabetes patients, which suggests that the potential disease population with mitochondrial disorders is greater than previously thought, and there may be many cases that have been misdiagnosed as cerebral infarction among the elderly. In the future, it is expected that the incidence of patients with cerebral infarction and DM will greatly increase. Thus, we should evaluate cerebral infarction in the elderly with caution to prevent the missed diagnoses of MELAS.
Acknowledgments
The authors would like to thank American Journal Experts (http://bit.ly/AJE-HS) for help with English language review.
Funding
No funding was obtained for the preparation of this case report.
Availability of Data and Materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author Contributions
Study concept and design: ST. Acquisition of data: OT. Analysis and interpretation of data: ST and OT. Drafting of the manuscript: ST. Critical revision of the manuscript for important intellectual content: all authors. All authors read and approved the final manuscript.
Conflict of Interests
The authors declare that they have no competing interests.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Abbreviations
mtDNA: mitochondrial DNA; MELAS: encephalomyopathy with lactic acidosis and stroke-like episodes; UTI: urinary tract infection; ADL: activities of daily living; CT: computed tomography; MRI: magnetic resonance imaging; ME: mitochondrial encephalomyopathy; RRF: ragged red fiber; DWI: diffusion-weighted imaging; FLAIR: fluid-attenuated inversion recovery; ADC: apparent diffusion coefficient
| References | ▴Top |
- Vanniarajan A, Nayak D, Reddy AG, Singh L, Thangaraj K. Clinical and genetic uniqueness in an individual with MELAS. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(5):440-444.
doi pubmed - Kadowaki T, Kadowaki H, Mori Y, Tobe K, Sakuta R, Suzuki Y, Tanabe Y, et al. A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N Engl J Med. 1994;330(14):962-968.
doi pubmed - Manwaring N, Jones MM, Wang JJ, Rochtchina E, Howard C, Mitchell P, Sue CM. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7(3):230-233.
doi pubmed - Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, DiMauro S, et al. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2(2):125-135.
doi - Aurangzeb S, Vale T, Tofaris G, Poulton J, Turner MR. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) in the older adult. Pract Neurol. 2014;14(6):432-436.
doi pubmed - Jones DL, Greenaway TM. Beware the thin, deaf 'type 2' diabetic: maternally inherited diabetes and deafness with systemic (mitochondrial) manifestations. Intern Med J. 2004;34(8):517-518.
doi pubmed - Koga Y, Povalko N, Nishioka J, Katayama K, Yatsuga S, Matsuishi T. Molecular pathology of MELAS and L-arginine effects. Biochim Biophys Acta. 2012;1820(5):608-614.
doi pubmed - Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, Kakuma T, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-624.
doi pubmed - Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651-653.
doi pubmed - Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, Simonetti S, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31(4):391-398.
doi pubmed - Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I, Ando M. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1994;35(4):413-419.
doi pubmed - King MP, Koga Y, Davidson M, Schon EA. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992;12(2):480-490.
doi pubmed - Kimata KG, Gordan L, Ajax ET, Davis PH, Grabowski T. A case of late-onset MELAS. Arch Neurol. 1998;55(5):722-725.
doi pubmed - Sharfstein SR, Gordon MF, Libman RB, Malkin ES. Adult-onset MELAS presenting as herpes encephalitis. Arch Neurol. 1999;56(2):241-243.
doi pubmed - Kisanuki YY, Gruis KL, Smith TL, Brown DL. Late-onset mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes with bitemporal lesions. Arch Neurol. 2006;63(8):1200-1201.
doi pubmed - Allard JC, Tilak S, Carter AP. CT and MR of MELAS syndrome. AJNR Am J Neuroradiol. 1988;9(6):1234-1238.
pubmed - Ito H, Mori K, Kagami S. Neuroimaging of stroke-like episodes in MELAS. Brain Dev. 2011;33(4):283-288.
doi pubmed - Kim JT, Lee YJ, Lee YM, Kang HC, Lee JS, Kim HD. Clinical characteristics of patients with non-specific and non-categorized mitochondrial diseases. Acta Paediatr. 2009;98(11):1825-1829.
doi pubmed - Scaglia F, Wong LJ, Vladutiu GD, Hunter JV. Predominant cerebellar volume loss as a neuroradiologic feature of pediatric respiratory chain defects. AJNR Am J Neuroradiol. 2005;26(7):1675-1680.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.