

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 9, Number 7, July 2017, pages 654-658
Gastrointestinal Tract Amyloidosis Presenting With Pneumatosis Intestinalis
Vikram Raghunathana, b, David Louisa, Baldeep Wirka
aDepartment of Medicine, Brown University, 593 Eddy Street, Providence, RI 02903, USA
bCorresponding Author: Vikram Raghunathan, Department of Medicine, Brown University, 593 Eddy Street, Providence, RI 02903, USA
Manuscript accepted for publication April 10, 2017
Short title: Pneumatosis Intestinalis From Amyloidosis
doi: https://doi.org/10.14740/jocmr2957w
| Abstract | ▴Top |
Pneumatosis intestinalis is a radiographic finding of gas pockets within the bowel wall. It can be associated with a range of diagnoses, but the most life-threatening causes are mesenteric ischemia, bowel necrosis, and bowel obstruction. Here we present the case of a patient with multiple myeloma who had pneumatosis intestinalis due to gastrointestinal amyloidosis, which is a rare manifestation of systemic amyloid disease. The patient had both transthyretin (ATTR) amyloidosis and acquired apolipoprotein serum amyloid A (AA) amyloidosis that are not usually seen in conjunction with multiple myeloma, which is most commonly associated with light-chain (AL) amyloidosis. This case highlights the importance of considering Congo red staining of bowel biopsies for amyloid deposition in patients undergoing endoscopy for unexplained gastrointestinal tract symptoms and even pneumatosis intestinalis, so as to avoid a delay in diagnosis that is typically seen with amyloidosis. Since each subtype of amyloidosis requires different therapy, amyloid subtyping is crucial, even with co-existing multiple myeloma.
Keywords: Multiple myeloma; Pneumatosis intestinalis; Amyloidosis
| Introduction | ▴Top |
Amyloidosis is a protein conformational disease from the extracellular deposition of misfolded aggregates of autologous proteins as non-branching fibrils in an anti-parallel twisted, beta-pleated sheet configuration [1]. These cross beta-pleated sheets accumulate systemically or in specific organs, impairing tissue function. Amyloidosis can be acquired or hereditary. More than 30 different proteins can cause amyloidosis [2]. The nomenclature includes the first letter A to specify amyloidosis followed by the precursor protein. The most common forms of systemic amyloidosis are: acquired monoclonal immunoglobulin light chain amyloidosis (AL) in 78% of cases, hereditary mutated transthyretin (ATTRm) amyloidosis in 7% of cases, acquired age-related wild-type transthyretin (ATTRwt) amyloidosis in 6%, and acquired apolipoprotein serum amyloid A (AA) amyloidosis in 6%. The remainder are rare forms such as hereditary mutated apolipoprotein A1 (AAapoA1) amyloidosis and hereditary lysozyme (ALys) amyloidosis [1-3].
Systemic AL amyloidosis (immunoglobulin light chain) is caused by the deposition of lambda (λ) or kappa (κ) light chains and can be primary or secondary. Secondary AL amyloidosis is associated with plasma cell dyscrasias, such as multiple myeloma in 30% of cases [4]. Amyloidosis has numerous clinical manifestations. The most common organs involved by AL amyloidosis are the heart (75%), kidney (proteinuria and renal failure, 65%), soft tissues (15%), liver (15%), peripheral and/or autonomic nervous system (10%), and gastrointestinal tract (GIT, 5%) [1]. AA amyloidosis involves the deposition of serum amyloid A, an acute phase reactant, and is seen in chronic inflammatory states. The most common organs infiltrated by AA amyloidosis are the kidney (proteinuria and renal failure, > 95%), liver (15%), heart (10%), and GIT (5%) [1]. Age-related ATTRwt amyloidosis can involve the heart (100% of cases presenting as diastolic heart failure), ligaments, and tenosynovium [1]. Hereditary mutated transthyretin ATTRm amyloidosis affects the peripheral and autonomic nervous system, heart, and eye [1].
GIT amyloidosis, caused by the accumulation of insoluble amyloid fibrils within the intestinal tissue, is a relatively uncommon complication of systemic amyloid disease. In a retrospective study of 2,334 amyloid patients over a 13-year period, Cowan et al found that only 76 patients (3.2%) had biopsy-proven GIT disease [3]. Of these 76 patients, 60 patients (79%) had systemic amyloidosis with dominant GIT involvement, whereas 16 patients (21%) had localized disease in the GIT [3].
GIT amyloidosis presents with bleeding, malabsorption, protein-losing enteropathy, and dysmotility [5]. Patients can have melena, hematochezia, esophageal reflux, diarrhea, constipation, nausea, abdominal pain, weight loss or early satiety [1]. The symptoms are due to direct amyloid fibril deposition in the bowel wall as well as secondary complications, including autonomic neuropathy, small bowel bacterial overgrowth, and cardiac cachexia [3].
Rarely, GIT amyloidosis can be complicated by pneumatosis intestinalis, which is most often detected radiographically as gas pockets within the bowel wall of the small or large intestine or both [6]. The clinical course of pneumatosis intestinalis from amyloidosis is not well characterized. Here we present an interesting case of pneumatosis intestinalis secondary to mixed AA and ATTR amyloidosis.
| Case Report | ▴Top |
An 86-year-old male with a history of lambda light chain multiple myeloma stage II (International Staging System), coronary artery disease, heart failure with preserved left ejection fraction, and hypertension presented to the emergency department with abdominal pain and distension.
Two months earlier, while undergoing evaluation of a left lower extremity deep vein thrombosis, he was found to have numerous lytic bone lesions of the ribs, spine, and pelvis. Computerized tomography (CT)-guided needle biopsy of a right tenth rib lesion showed dense infiltration with monomorphic plasma cells. The serum protein electrophoresis was negative, but the serum protein immunofixation electrophoresis showed lambda light chains. The urine protein electrophoresis was negative, but the urine protein immunofixation electrophoresis showed lambda light chains. Beta-2-microglobulin was 4.66 mg/L (0.7 - 2.5 mg/L) and albumin was 4 g/dL (3.5 - 5.5 g/dL). The serum free kappa light chains were 9.5 mg/L (3.3 - 19.4 mg/L), serum free lambda light chains were 15,300 mg/L (5.7 - 26.3 mg/L) and the kappa-lambda ratio was close to 0 (0.26 - 1.65). The calcium was normal. He was diagnosed with stage II lambda light chain multiple myeloma and treatment with lenalidomide, bortezomib, and dexamethasone chemotherapy was initiated.
On admission, the patient reported abdominal pain, distention, and constipation beginning shortly after his second cycle of chemotherapy the previous week. He denied nausea or emesis. On physical exam, he was afebrile and hypotensive with a blood pressure of 90/41. He had a moderately distended, non-tender, tympanitic abdomen with absent bowel sounds. There was no rebound tenderness, rigidity, or guarding on abdominal exam.
Laboratory values were notable for white blood cell count 6.4 × 109/L (3.5 - 11 × 109/L), hemoglobin 8.6 g/dL (13.5 - 16 g/dL), platelets 15 × 109/L (150 - 400 × 109/L), serum creatinine 2.35 mg/dL (0.64 - 1.37 mg/dL), calcium 9.9 mg/dL (8.8 - 10.5 mg/dL), albumin 3.9 g/dL (3.5 - 5.5 g/dL), lactate 1 mEq/L (0.3 - 2.2 mEq/L), and spot urine protein 30 mg/dL (normal < 10 mg/dL). The troponin T level was < 0.01 ng/mL. The B-type natriuretic peptide was 950 pg/mL (0 - 176 pg/mL). Electrocardiogram showed normal sinus rhythm. Echocardiography revealed left ventricular ejection fraction of 60% and interventricular septal thickness of 8 mm. Abdominal CT scan showed diffuse colonic distension with submucosal gas pockets and numerous polypoid mucosal lesions with bowel wall thickening but no portal venous air or free intra-abdominal air (Fig. 1). There was no whirl sign.
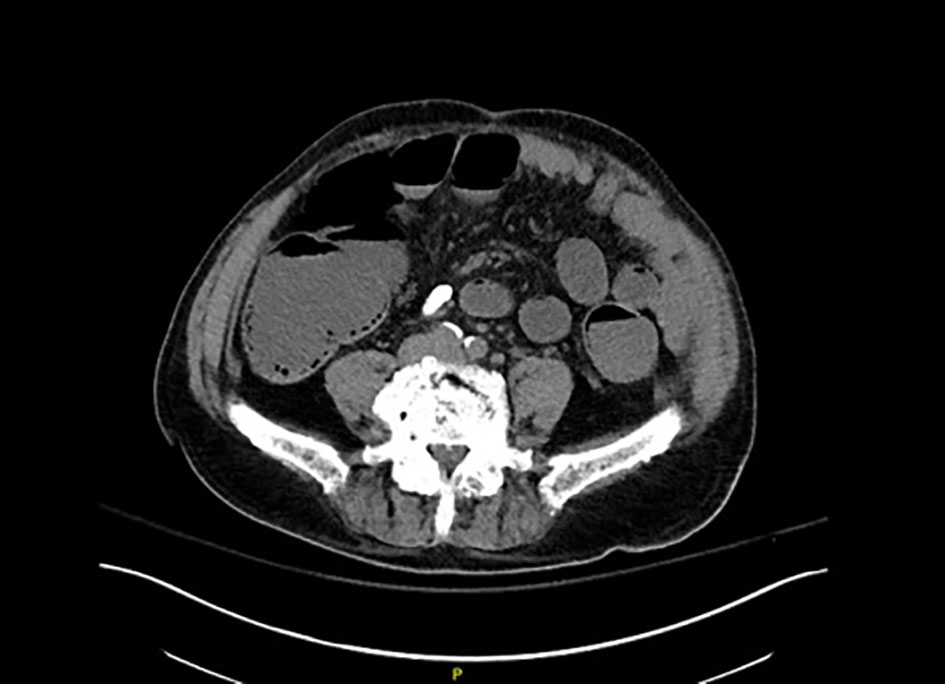 Click for large image | Figure 1. Non-contrast abdominal CT demonstrating widespread colonic distension and air-fluid levels along with numerous areas of submucosal gas in the colonic wall. |
The patient was admitted to the hematology-oncology service and treated with intravenous fluids, ciprofloxacin, and metronidazole for suspected infectious colitis. Soon thereafter, he developed profuse, watery diarrhea and persistent hypotension. Blood cultures, stool bacterial cultures, and Clostridium difficile toxin were negative. Empiric antimicrobial coverage was continued, and due to worsening hypotension, the patient was transferred to the intensive care unit. Further investigation included a colonoscopy.
Colonoscopy revealed submucosal polypoid lesions involving the transverse, descending, and sigmoid colon (Fig. 2). Colonic biopsy showed submucosal eosinophilic material with apple-green birefringence under polarized light when stained with Congo red dye, confirming the diagnosis of amyloidosis. Immunohistochemistry of the colon biopsy showed strong signals for AA and ATTR amyloidosis (Fig. 3). Antibiotics were discontinued, and an antimotility bowel regimen was started leading to the resolution of his symptoms. He was discharged home and continued chemotherapy for the multiple myeloma as an outpatient.
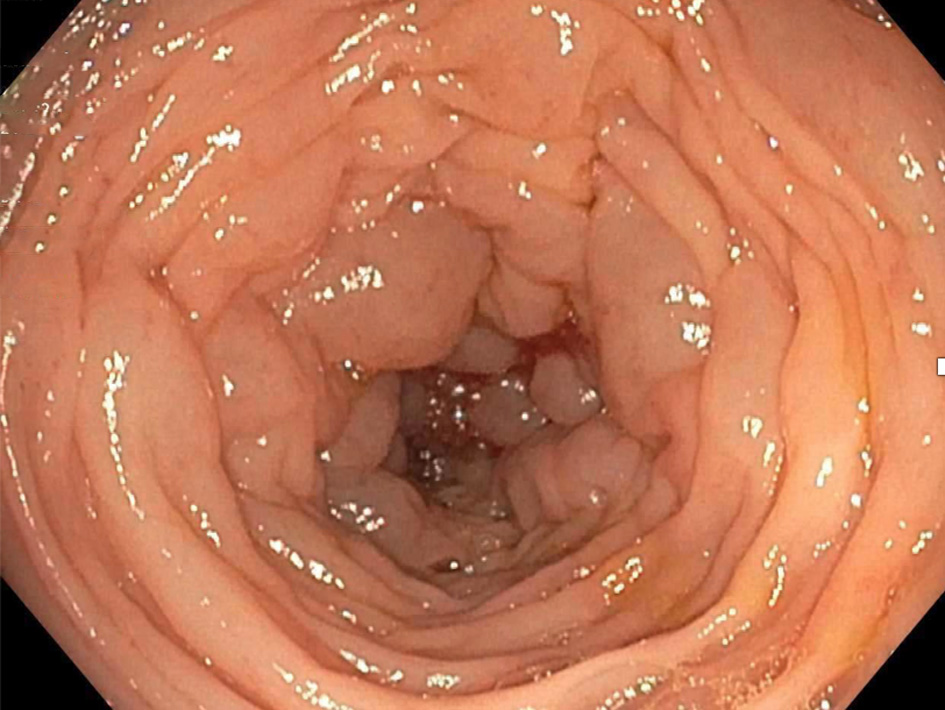 Click for large image | Figure 2. Colonoscopy image showing submucosal polypoid lesions of the descending colon. |
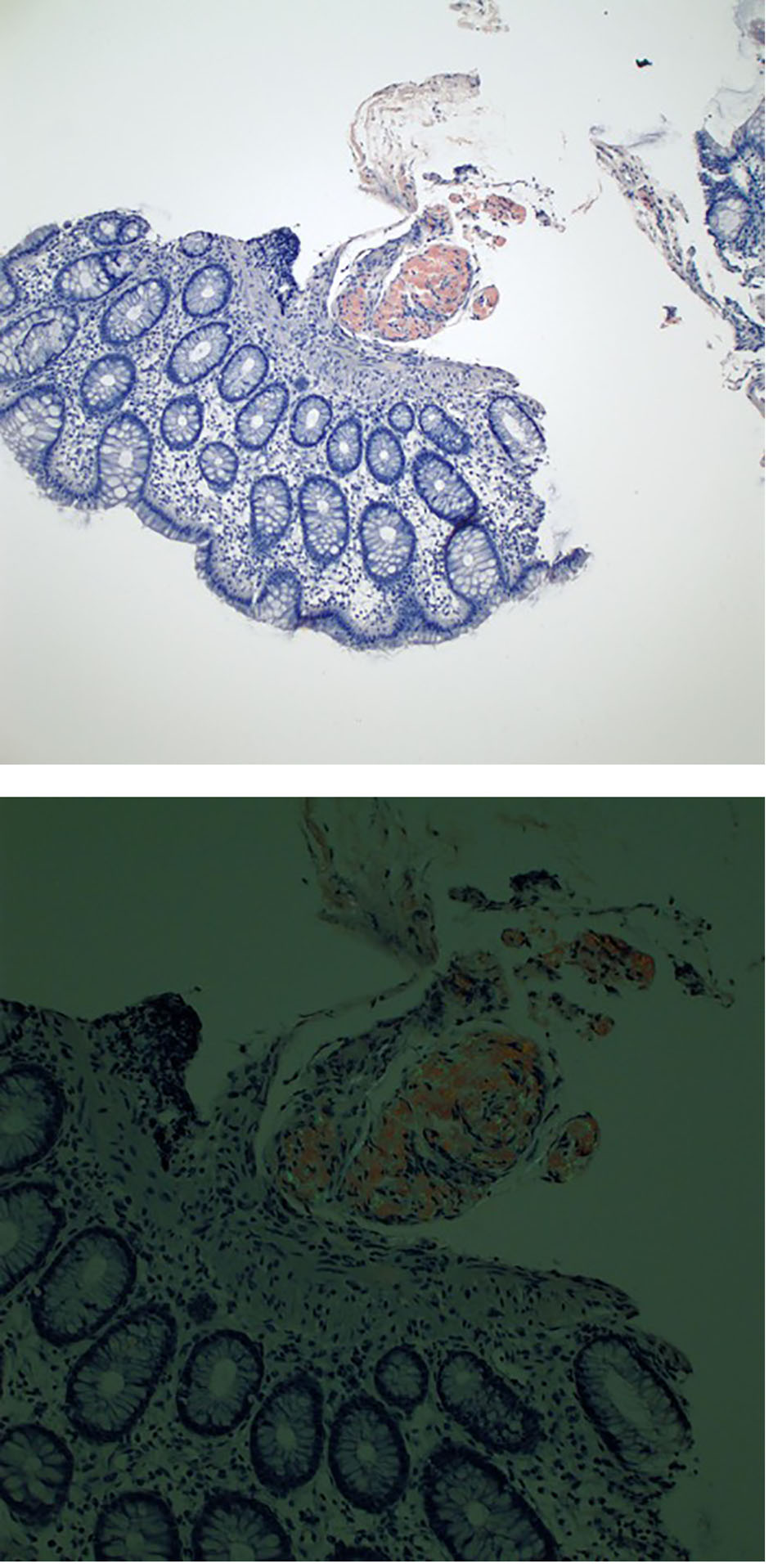 Click for large image | Figure 3. Congo red stain of colon tissue showing amyloid deposits within the submucosa (top) and illustrating apple-green birefringence under polarized light (bottom). |
| Discussion | ▴Top |
Pneumatosis intestinalis is a radiographic finding and is characterized by gas pockets within the wall of the small or large bowel [6]. There are more than 60 causes of pneumatosis intestinalis [6]. The most life-threatening causes are mesenteric ischemia, bowel necrosis, and bowel obstruction. Cases of gastrointestinal amyloidosis presenting with pneumatosis intestinalis are very uncommon. The pathogenesis of pneumatosis intestinalis is unknown but is postulated to be due to disruption of the intestinal mucosal integrity by amyloid deposits, allowing for infiltration of luminal gas or gas-producing bacteria [6].
There are no large prospective, randomized studies of pneumatosis intestinalis in patients with amyloidosis although individual cases have been described (Table 1) [5, 7, 8]. Pearson et al described a 76-year-old male with IgG kappa multiple myeloma who presented with abdominal pain and had radiographic findings of pneumatosis intestinalis, hepatic portal venous gas, and small bowel obstruction [7]. At laparotomy, a 4-cm segment of small bowel was resected at the obstruction site. Pathology revealed air-filled cysts in the small bowel wall and infiltration of the muscularis propria by eosinophilic material. Congo red staining showed apple-green birefringence under polarized light consistent with amyloidosis [7]. Sub-typing of the amyloid deposits was not performed. The patient died 15 months after diagnosis from recurrent small bowel obstruction [7]. Yamamoto et al described a 63-year-old female with rheumatoid arthritis who had pneumatosis intestinalis and hepatic portal venous gas due to AA amyloidosis [8]. The patient was treated with conservative management [8]. Similarly, Khalid et al described a 46-year-old male with melena, pneumoperitoneum, and extensive pneumatosis intestinalis [5]. He had an exploratory laparotomy, and no free fluid or bowel contents were found. In this case, the free air under the diaphragm was not a true bowel perforation but rather a rupture of subserosal air cysts. An esophagogastroduodenoscopy and small bowel biopsy showed amyloidosis by Congo red stain [5]. Sub-typing of the amyloid was not performed. The patient recovered with conservative management. Given the rarity of amyloidosis presenting with pneumatosis intestinalis, there are insufficient data regarding prognosis and long-term clinical outcomes.
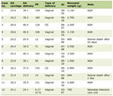 Click to view | Table 1. Characteristics of Patients With Pneumatosis Intestinalis From Amyloidosis |
The radiographic finding of pneumatosis intestinalis without concurrent signs of bowel obstruction, mesenteric ischemia, portal venous gas, or free intra-abdominal air can be managed medically by treating the underlying cause [6]. However, in cases with the whirl sign and pneumatosis intestinalis, suggestive of fulminant bowel infarction by intestinal volvulus, there is a 25-fold increase rate of the need for surgery [9]. CT has the best sensitivity at diagnosing pneumatosis intestinalis and can detect portal venous gas and as little as 1 mL of pneumoperitoneum [5].
Importantly, in cases from the medical literature as well as ours, the diagnosis of GIT amyloidosis would not have been made in the absence of Congo red staining of bowel tissue. Given the many causes of pneumatosis intestinalis, testing for amyloidosis may not ordinarily be pursued [6]. This is particularly significant in light of evidence that patients with amyloidosis often face delayed diagnosis due to their non-specific symptoms. In a study of 919 patients with pneumatosis intestinalis, the most common symptoms in descending order were: diarrhea, melena, abdominal pain, constipation, weight loss, and tenesmus [10]. Cowan et al found that patients with systemic AL amyloidosis, the most common form, had a median of 5 months between onset of symptoms and diagnosis, whereas patients with ATTR amyloidosis faced an 8-month lag [3]. In contrast, patients who present with peripheral neuropathy have a 48-month delay in the diagnosis of amyloidosis [11]. In our case, there was a month between the onset of GIT symptoms and diagnosis of amyloidosis. The GIT is amenable to biopsy, allowing for a more timely diagnosis. Tada et al found that the most common site of GIT involvement with amyloidosis was the small intestine (especially the second part of the duodenum) followed by the stomach and colorectum [12].
Even though the different types of systemic amyloidosis have overlapping clinical manifestations, each type of amyloidosis requires different therapy. For example, AL amyloidosis is treated with anti-plasma cell chemotherapy and autologous peripheral blood stem cell transplantation [1]. However, patients with active bleeding from GIT AL amyloidosis can have catastrophic gastrointestinal hemorrhage from a myeloablative autologous peripheral blood stem cell transplant [3]. On the other hand, ATTR amyloidosis, the most common systemic type of non-AL amyloidosis, is treated with agents such as doxycycline or epigallocatechin gallate, which interfere with the aggregation of transthyretin [1]. The principle of therapy for AA amyloidosis is to treat the underlying cause. For example, a case of GIT AA amyloidosis secondary to rheumatoid arthritis resolved with tocilizumab (humanized anti-interleukin-6 receptor antibody) therapy for the rheumatoid arthritis [13]. Therefore, sub-typing of the amyloid deposits is crucial. For example, immunohistochemistry with commercial antibodies can accurately sub-type amyloidosis in more than 95% of cases [14]. Immunoelectron microscopy with commercial antibodies can accurately sub-type more than 99% of amyloidosis cases [15]. Other methods include mass spectrometry and gene sequencing [1].
Amyloid sub-typing in our patient revealed mixed AA/ATTR amyloidosis, an uncommon and unexpected finding. Common clinical findings of secondary AL amyloidosis from multiple myeloma are congestive heart failure, edema (from protein-wasting nephropathy or renal failure), glossomegaly, and periorbital ecchymosis. AL amyloidosis is also the most common type associated with gastrointestinal involvement. Cowan et al found that 84% of patients with GIT-dominant amyloidosis had AL amyloidosis, 8% had ALys amyloidosis, and 8% had ATTR amyloidosis [3]. In addition, ATTR amyloidosis is associated with diastolic heart failure in the majority of cases, as possibly in our patient’s case as well [1].
Our patient demonstrates that Congo red staining of bowel biopsies in patients undergoing endoscopy should be considered for those with unexplained chronic gastrointestinal symptoms and even pneumatosis intestinalis. Cases of amyloidosis may be overlooked if patients present primarily with gastrointestinal symptoms. Crucially, multiple myeloma may coexist with non-light chain forms of amyloidosis. The initial assumption in multiple myeloma patients who are found to have amyloidosis is that they have AL amyloidosis. However, our patient with multiple myeloma was found to have AA amyloidosis and ATTR amyloidosis, which will not respond to the standard treatments for AL amyloidosis. Therefore, sub-typing of the amyloid is vital in order for the correct treatment to be given.
Abbreviations
KUB: kidney, ureter, and bladder plain film X-ray; CT: computed tomography; GIT: gastrointestinal tract.
| References | ▴Top |
- Palladini G, Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood. 2016;128(2):159-168.
doi pubmed - Ankarcrona M, Winblad B, Monteiro C, Fearns C, Powers ET, Johansson J, Westermark GT, et al. Current and future treatment of amyloid diseases. J Intern Med. 2016;280(2):177-202.
doi pubmed - Cowan AJ, Skinner M, Seldin DC, Berk JL, Lichtenstein DR, O'Hara CJ, Doros G, et al. Amyloidosis of the gastrointestinal tract: a 13-year, single-center, referral experience. Haematologica. 2013;98(1):141-146.
doi pubmed - Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45-59.
pubmed - Khalid F, Kaiyasah H, Binfadil W, Majid M, Hazim W, ElTayeb Y. Pneumatosis intestinalis due to gastrointestinal amyloidosis: A case report & review of literature. Int J Surg Case Rep. 2016;23:29-32.
doi pubmed - Khalil PN, Huber-Wagner S, Ladurner R, Kleespies A, Siebeck M, Mutschler W, Hallfeldt K, et al. Natural history, clinical pattern, and surgical considerations of pneumatosis intestinalis. Eur J Med Res. 2009;14(6):231-239.
doi pubmed - Pearson DC, Price LM, Urbanski S. Pneumatosis cystoides intestinalis: an unusual complication of systemic amyloidosis. J Clin Gastroenterol. 1996;22(1):74-76.
doi pubmed - Yamamoto A, Kawahito Y, Niimi M, Kadoya M, Hamaguchi M, Ishino H, Wada M, et al. Pneumatosis intestinalis and hepatic portal venous gas caused by gastrointestinal perforation with amyloidosis. J Clin Gastroenterol. 2008;42(6):758-759.
doi pubmed - Duda JB, Bhatt S, Dogra VS. Utility of CT whirl sign in guiding management of small-bowel obstruction. AJR Am J Roentgenol. 2008;191(3):743-747.
doi pubmed - Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
- Duston MA, Skinner M, Anderson J, Cohen AS. Peripheral neuropathy as an early marker of AL amyloidosis. Arch Intern Med. 1989;149(2):358-360.
doi pubmed - Tada S, Iida M, Iwashita A, Matsui T, Fuchigami T, Yamamoto T, Yao T, et al. Endoscopic and biopsy findings of the upper digestive tract in patients with amyloidosis. Gastrointest Endosc. 1990;36(1):10-14.
doi - Inoue D, Arima H, Kawanami C, Takiuchi Y, Nagano S, Kimura T, Shimoji S, et al. Excellent therapeutic effect of tocilizumab on intestinal amyloid a deposition secondary to active rheumatoid arthritis. Clin Rheumatol. 2010;29(10):1195-1197.
doi pubmed - Satoskar AA, Efebera Y, Hasan A, Brodsky S, Nadasdy G, Dogan A, Nadasdy T. Strong transthyretin immunostaining: potential pitfall in cardiac amyloid typing. Am J Surg Pathol. 2011;35(11):1685-1690.
doi pubmed - Fernandez de Larrea C, Verga L, Morbini P, Klersy C, Lavatelli F, Foli A, Obici L, et al. A practical approach to the diagnosis of systemic amyloidoses. Blood. 2015;125(14):2239-2244.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.