

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 8, Number 11, November 2016, pages 819-823
Generalized Lymphadenopathy as Presenting Feature of Systemic Lupus Erythematosus: Case Report and Review of the Literature
Wais Afzala, c, Talal Araba, Tofura Ullahb, Katerina Tellera, Kaushik J. Doshia
aDepartment of Internal Medicine, Jamaica Hospital Medical Center, 8900 Van Wyck Expressway, Jamaica, NY 11418, USA
bDepartment of Clinical Research, Jamaica Hospital Medical Center, 8900 Van Wyck Expressway, Jamaica, NY 11418, USA
cCorresponding Author: Wais Afzal, Department of Internal Medicine, Jamaica Hospital Medical Center, 8900 Van Wyck Expressway, Jamaica, NY 11418, USA
Manuscript accepted for publication September 02, 2016
Short title: Lupus Lymphadenopathy
doi: http://dx.doi.org/10.14740/jocmr2717w
| Abstract | ▴Top |
Lymphadenopathy could represent a vast spectrum of etiologies including infectious and non-infectious diseases. Besides proper history taking, physical examination, and laboratory investigations, a tissue diagnosis is often necessary to unmask the cause of generalized lymphadenopathy. Here we present a 23-year-old woman who was admitted for diffuse generalized lymphadenopathy, fatigue, malaise, weight loss, nausea, and bilateral lower extremity edema. She reported a history of seizures as well as stroke 2 years prior with no other medical conditions present. Although malignant and infectious etiologies were initially the primary targets for workup, her history of seizures and stroke remained a dilemma. Extensive workup for malignant and infectious diseases was unrevealing; however, rheumatologic workup was eventually positive for systemic lupus erythematosus (SLE). This case illustrates that extensive generalized diffuse lymphadenopathy may be a presenting feature of SLE and should be considered in the differential diagnosis of patients presenting with diffuse lymphadenopathy and constitutional symptoms.
Keywords: Lymphadenopathy; Systemic lupus erythematosus; Seizure; Stroke
| Introduction | ▴Top |
Lymphadenopathy could be the presenting feature of many ailments including infectious or non-infectious diseases. Infectious etiologies include viral, bacterial, fungal, parasitic, and rickettsial causes. Common causes of non-infectious lymphadenopathy include malignancy, sarcoidosis, connective tissue disease, and Kikuchi-Fujimoto disease. The initial approach to discern the cause of lymphadenopathy begins with proper history and physical examination followed by laboratory investigations. A tissue diagnosis is often necessary to unmask the cause of lymphadenopathy.
Systemic lupus erythematosus (SLE) is a chronic multisystem autoimmune inflammatory disease with diverse clinical manifestations. It presents most often in young females. The diagnosis of SLE is established by the presence of four or more of the 11 American College of Rheumatology (ACR) criteria, none of which include lymphadenopathy. The precise prevalence of lymphadenopathy in SLE is unknown. There are several case reports suggesting that generalized lymphadenopathy could be the first manifestation of SLE [1-4]. Here we present a 23-year-old female who came to our hospital with diffuse generalized lymphadenopathy, fatigue, malaise, weight loss, nausea, and bilateral lower extremity edema. She underwent extensive workup for malignant, infectious, and immunologic diseases and was ultimately diagnosed with SLE. This case adds to the body of evidence suggesting that diffuse generalized lymphadenopathy may be the presenting feature of SLE. It should be considered in the differential diagnosis of patients presenting with diffuse lymphadenopathy and constitutional symptoms in select groups of patients, such as young women.
| Case Report | ▴Top |
A 23-year-old Afro-Jamaican woman presented with generalized lymphadenopathy of 7-8 months duration. She complained of gradual worsening fatigue, malaise, unintentional weight loss of 40 pounds, nausea, intermittent headaches, and progressively worsening bilateral lower extremity edema. She denied fever, chills, night sweats, loss of appetite, rash, sore throat, cough, and shortness of breath. At age 15, she had an episode of left facial palsy which lasted 7 months and resolved spontaneously. She experienced tonic-clonic seizures 2 years ago and had been on sodium valproate 200 mg daily since that time. Her last episode of seizures was 3 months ago. Two years prior, she suffered from ischemic stroke with residual left-sided weakness. She was never diagnosed with infectious diseases, sarcoidosis, or malignancies and had no family history of such. The patient lived most of her life in the Jamaican Republic and was visiting the United States.
Physical examination revealed a well-developed, well-nourished young female in no acute distress with normal vital signs. Heart and lung examinations were unremarkable. Abdominal examination was consistent with hepatomegaly and mild splenomegaly. There was 2+ pitting edema of lower extremities up to shins left more than right. There were multiple painless, enlarged, soft, non-tender, freely movable lymph nodes in the cervical, axillary, and inguinal areas, the largest of which measured 5 × 4 cm. On neurological examination, she was found to have left facial droop, motor strength of 5/5 on right upper and lower extremities, and 3/5 on left upper and lower extremities. There was decreased sensation on the left side, and slow finger to nose on the left. She was limping and used a cane for ambulation. Her neurologic signs and symptoms were the sequelae of the ischemic stroke 2 years prior. She was admitted with possible diagnosis of malignancy versus infectious etiology. Nevertheless, the history of stroke in this young woman with no known cardiovascular risk factors remained a dilemma. Initial investigations are presented in Table 1.
 Click to view | Table 1. Initial Investigations |
Considering diffuse lymphadenopathy on physical examination and pancytopenia on laboratory investigation, malignancy in particular lymphoma was considered the most likely diagnosis. Moreover, her hypoalbuminemia, proteinuria, and hyperlipidemia were concerning for possible nephrotic syndrome. A computed tomography (CT) scan of the head without contrast revealed an old infarct and encephalomalacia in the right posterior insula, right superior temporal gyrus and adjacent to parietal lobe, and old infarcts in the bilateral frontal lobes and left parietal lobe. Numerous enlarged cervical lymph nodes (Fig. 1a) with hypertrophy of nasopharyngeal soft tissues and palatine and lingual tonsils were seen. A CT of the chest with IV contrast showed bilateral axillary lymphadenopathy (Fig. 1b). Mediastinal and right hilar lymph nodes were also enlarged. CT scan of abdomen and pelvis with IV contrast indicated hepatomegaly (17.7 cm in length), mild splenomegaly (12.9 cm) anterior-posteriorly, a hypodense lesion in the periphery of the spleen (1.4 × 3.4 × 3.6 cm), as well as posterior splenic contour nodularity (Fig. 1c). There was periportal, mesenteric, retroperitoneal, common iliac, pelvic sidewall, external iliac, and inguinal lymphadenopathy bilaterally as demonstrated by the CT scan of the abdomen and pelvis (Fig. 1d).
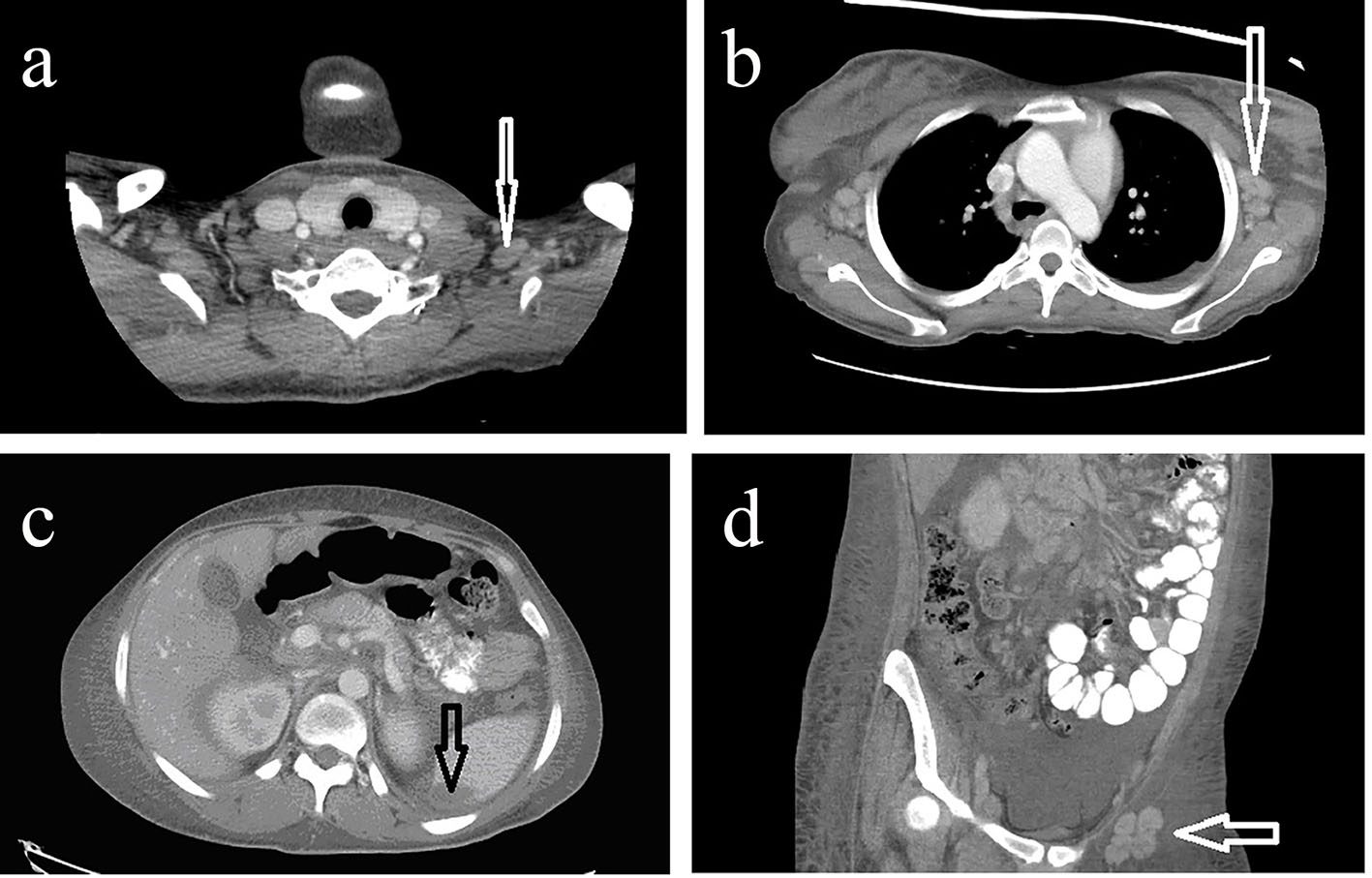 Click for large image | Figure 1. (a) CT neck with IV contrast demonstrating multiple enlarged supraclavicular lymph nodes. (b) CT chest with IV contrast demonstrating enlarged axillary lymph nodes. (c) CT abdomen and pelvis with IV contrast demonstrating a hypodense lesion in the periphery of the spleen measuring 1.4 × 3.4 × 3.6 cm. (d) CT abdomen and pelvis with IV contrast demonstrating multiple enlarged external iliac lymph nodes. |
Excisional lymph node biopsy was performed from the right inguinal area. The results of the biopsy were reported as reactive lymph node with follicular hyperplastic pattern and no evidence of lymphoma (Fig. 2a and b). Flow cytometry and marker results did not support malignancy. Acid-fast stain, bacterial, mycobacterial and fungal cultures were all reported as negative.
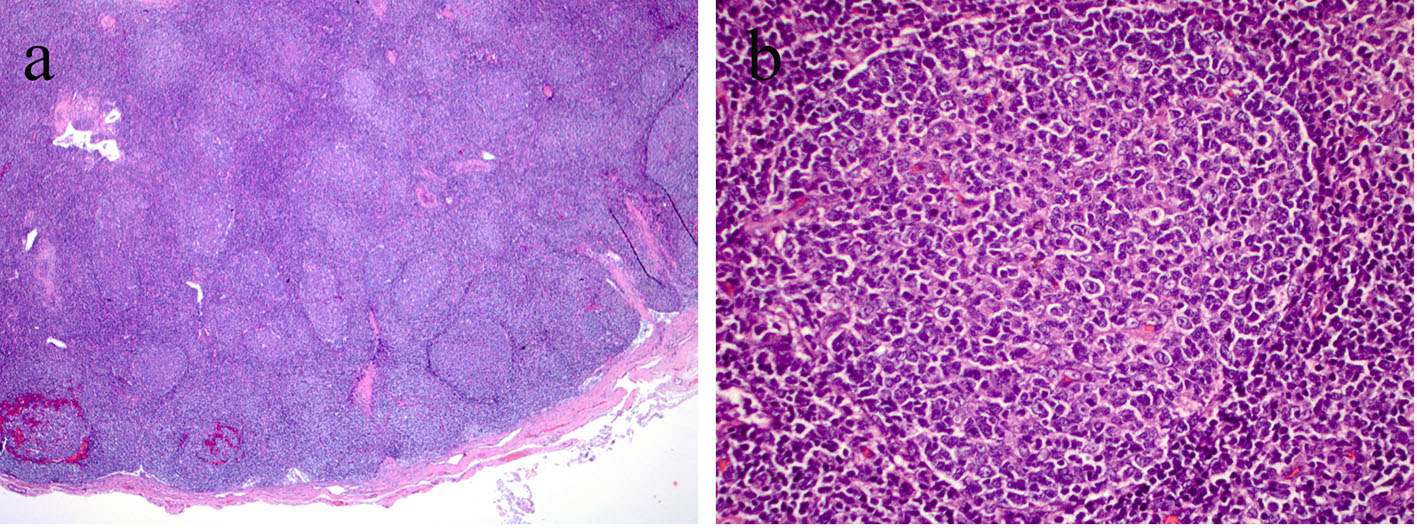 Click for large image | Figure 2. (a) Right inguinal lymph node sample indicating numerous hyperplasic follicles (H&E, × 10 magnification). (b) Right inguinal lymph node sample indicating hyperplastic follicles with prominent germinal centers and reactive lymphocytosis (H&E, × 40 magnification). |
Further investigations for human immunodeficiency virus (HIV 1 and 2), heterophile antibody, human T-cell lymphotropic virus (HTLV 1 and 2) antibodies, and purified protein derivative (PPD) were all negative. Anticardiolipin IgA and IgM antibodies were positive; however, anticardiolipin IgG was reported as negative. Antinuclear antibodies (ANA) were also positive with high titer of 1:1,280.
Owing to proteinuria of ≥ 500 mg/dL on urine analysis, a 24-h urine protein was performed and found to be 4,338 mg/24 h. This was consistent with nephrotic syndrome. A complete panel of lupus serology was sent, and diagnosis of active lupus was confirmed (Table 2). Pulse dose of methylprednisolone 1 g IV was started for 3 days and switched to oral prednisone 60 mg daily.
Click to view | Table 2. Connective Tissue Disease Panel |
Despite encouragement, the patient refused to proceed with renal biopsy. Neurology evaluation was completed to assess possible causes of previous stroke with magnetic resonance angiography (MRA) of the brain, carotid Doppler, and transthoracic echocardiogram (TTE) with bubble study. MRA revealed multiple chronic small cerebral infarcts more on the right than left hemispheres and narrowing of right more than left middle cerebral arteries. Carotid arteries were less than 30% stenotic bilaterally suggesting lupus vasculitis as the possible cause of the ischemic stroke. The patient was discharged on prednisone 60 mg oral daily tapering dose, calcium and vitamin D supplement, aspirin 81 mg oral daily, lisinopril 2.5 mg oral daily, and atorvastatin 80 mg oral at night. She was also advised to continue sodium valproate and follow with rheumatology and nephrology clinics. The plan was to start mycophenolate mofetil and hydroxychloroquine and tapper off oral prednisone. Unfortunately, the patient did not follow with rheumatology clinic and was lost to follow-up.
| Discussion | ▴Top |
Lymphadenopathy could represent a vast spectrum of etiologies including infectious or non-infectious diseases. Some of the infectious causes include HIV, Castleman’s disease, tuberculosis, brucellosis, and syphilis. Common causes of non-infectious etiologies of diffuse lymphadenopathy include lymphoma, sarcoidosis, connective tissue disease, and Kikuchi-Fujimoto disease. The initial approach to delineate the cause of lymphadenopathy begins with obtaining proper history and physical examination. Laboratory investigations focusing on infections, malignancies, and immunological causes are of paramount importance and can lead to a proper diagnosis. Tissue diagnosis is often needed to unmask the cause of lymphadenopathy. Large-scale studies advocate for excisional biopsy as the gold standard method with minimal morbidity and mortality for rectifying benign and malignant causes of lymphadenopathy [5].
SLE is a chronic multisystem autoimmune inflammatory disease with diverse clinical manifestations. It most often affects young females. According to Systemic Lupus International Collaborating Clinics (SLICC) classification criteria, generalized lymphadenopathy is not considered a criterion for diagnosis [6]. The prevalence of generalized lymphadenopathy is by far less as seen in SLE patients previously [1]. The precise prevalence of lymphadenopathy in SLE patient is unknown. In one retrospective study of 90 SLE patients, 23 patients (26%) were found to have lymphadenopathy [7]. The same study indicated that SLE patients presenting with lymphadenopathy are more likely to manifest constitutional symptoms such as fever, fatigue, weight loss, cutaneous symptoms and signs, higher rates of hepatomegaly and splenomegaly, increased anti-dsDNA antibodies, and decreased complement levels. This illustrates that lymphadenopathy is indicative of active disease. There are several case reports suggesting that generalized lymphadenopathy could be the first manifestation of SLE [1-4].
In the patient described in this report, all of these clinical features were seen with the exception of cutaneous manifestations. There was no history of rash, photosensitivity, oral ulcers, arthritis, or serositis. The diagnosis of SLE was established by ruling out other more common causes of lymphadenopathy, followed by her history of seizures, documented nephrotic range proteinuria, pancytopenia, positive high titers of ANA, anti-dsDNA, anti-SM, low C3 and C4, and the presence of anticardiolipin IgM antibodies.
At the histologic level, manifestations of SLE lymphadenopathy are non-specific and include follicular hyperplasia, scattered immunoblasts, and plasma cells with increased vascularity [8]. Biopsy result in our patient yielded similar findings. Lymph node biopsy, in addition to flow cytometry and markers, can exclude lymphoma as a cause of lymphadenopathy. Since our patient did not consent for renal biopsy, the degree of renal involvement could not be staged properly. Though lymphoma may not be an initial diagnosis, there is strong evidence demonstrating that patients with SLE are at increased risk of developing non-Hodgkin lymphoma, Hodgkin lymphoma, leukemia, and some non-hematologic malignancies [9]. Thus, a lymph node biopsy is crucial even if the diagnosis of SLE is confirmed.
Other diseases that need to be considered and ruled out in presence of generalized lymphadenopathy include sarcoidosis, Kikuchi-Fujimoto disease, and Castleman’s disease.
Sarcoidosis is a multisystem granulomatous disease with heterogeneous presentation characterized by cellular alteration in lung tissue and intrathoracic lymphadenopathy. Histopathologically, sarcoidosis causes widespread non-caseating granulomas, which is very specific to this disease. Elevated serum calcium is also seen. Sarcoidosis in this young woman with African roots was a possibility. However, absence of non-caseating granulomas on lymph node biopsy and normal serum calcium was not suggestive of this diagnosis.
Kikuchi-Fujimoto disease, also known as histiocytic necrotizing lymphadenitis, is an extremely rare disease with worldwide distribution with high predilection for young Asian females and it is known to have favorable prognosis. Common clinical manifestations include fever, chills, upper respiratory symptoms, and lymphadenopathy with cervical lymphadenopathy the most common [10]. The characteristic lymph node histopathologic features of Kikuchi-Fujimoto disease include irregular paracortical areas of coagulative necrosis with abundant karyorrhectic debris, and abundance of histiocytes at the margin of the necrotic areas [11]. Our patient’s lymph node biopsy did not have any of these features. There is evidence indicating Kikuchi-Fujimoto disease has a rare association with SLE, the diagnosis of which may precede, coincide or follow SLE [12].
Castleman’s disease, a rare lymphoproliferative disorder, has an equal predilection for both sexes and may affect anyone from adolescence to seventh decade of life [13]. Clinical manifestations include fever, fatigue, night sweats, anemia, and lymphadenopathy. The extent of lymphadenopathy depends on if the disease is unicentric or multicentric. The lymph node biopsy may reveal increased plasma cell proliferation within the follicles with increased vascularity [14]. Our patient’s lymph node biopsy did not have any of these features.
As discussed earlier, our patient was found to be negative for HIV-1 and 2, HTLV-1 and 2 antibodies, as well as infectious mononucleosis. Negative bacterial, fungal and mycobacterial cultures have effectively ruled out these as the cause. Testing for brucellosis and syphilis were never performed.
Conclusion
Lymphadenopathy could be seen in the course of SLE; however, it is not considered a diagnostic criterion for the disease. The prevalence of generalized lymphadenopathy in SLE is much less in comparison to cases encountered in the past. Evidence suggests that generalized lymphadenopathy could be indicative of active disease. In addition to proper history, physical examination, and laboratory investigations, a tissue diagnosis based on excisional lymph node biopsy is required to differentiate SLE lymphadenopathy from other diseases, particularly malignancy.
Acknowledgments
The authors are very thankful to Dr. Kam Newman at the National Institute of Arthritis and Musculoskeletal and Skin Diseases for the revision of the manuscript.
Disclosure
The manuscript, patient data, and imaging are reproduced in accordance with Jamaica Hospital Medical Center policies approved by internal IRB.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
| References | ▴Top |
- Smith LW, Gelber AC, Petri M. Diffuse lymphadenopathy as the presenting manifestation of systemic lupus erythematosus. J Clin Rheumatol. 2013;19(7):397-399.
doi pubmed - Kitsanou M, Andreopoulou E, Bai MK, Elisaf M, Drosos AA. Extensive lymphadenopathy as the first clinical manifestation in systemic lupus erythematosus. Lupus. 2000;9(2):140-143.
doi pubmed - Shrestha D, Dhakal AK, Shiva RK, Shakya A, Shah SC, Shakya H. Systemic lupus erythematosus and granulomatous lymphadenopathy. BMC Pediatr. 2013;13:179.
doi pubmed - Gillmore R, Sin WY. Systemic lupus erythematosus mimicking lymphoma: the relevance of the clinical background in interpreting imaging studies. BMJ Case Rep. 2014;2014.
- Ozkan EA, Goret CC, Ozdemir ZT, Yanik S, Goret NE, Dogan M, Cihan FG, et al. Evaluation of peripheral lymphadenopathy with excisional biopsy: six-year experience. Int J Clin Exp Pathol. 2015;8(11):15234-15239.
pubmed - Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686.
doi pubmed - Shapira Y, Weinberger A, Wysenbeek AJ. Lymphadenopathy in systemic lupus erythematosus. Prevalence and relation to disease manifestations. Clin Rheumatol. 1996;15(4):335-338.
doi pubmed - Papadaki HA, Xylouri I, Katrinakis G, Foudoulakis A, Kritikos HD, Stathopoulos EN, Boumpas DT, et al. Non-Hodgkin's lymphoma in patients with systemic lupus erythematosus. Leuk Lymphoma. 2003;44(2):275-279.
doi pubmed - Cao L, Tong H, Xu G, Liu P, Meng H, Wang J, Zhao X, et al. Systemic lupus erythematous and malignancy risk: a meta-analysis. PLoS One. 2015;10(4):e0122964.
doi pubmed - Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26(1):50-54.
doi pubmed - Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004;122(1):141-152.
doi pubmed - Santana A, Lessa B, Galrao L, Lima I, Santiago M. Kikuchi-Fujimoto's disease associated with systemic lupus erythematosus: case report and review of the literature. Clin Rheumatol. 2005;24(1):60-63.
doi pubmed - Barua A, Vachlas K, Milton R, Thorpe JA. Castleman's disease - a diagnostic dilemma. J Cardiothorac Surg. 2014;9:170.
doi pubmed - Casper C. The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol. 2005;129(1):3-17.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.