

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 8, Number 4, April 2016, pages 317-324
Efficacy of Stem Cell Therapy in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis
Mirian Conceicao Mouraa, e, Maria Rita Carvalho Garbi Novaesb, Yuri S. S. P. Zagoc, Emanoel Junio Eduardoc, Luiz Augusto Casularid
aHospital Regional da Asa Norte, State Secretariat of Health of the Federal District, DF, Brazil
bSchool of Health Sciences, DF, and University of Brasilia, Brazil
cSchool of Medicine, School of Health Sciences, DF, Brazil
dUniversity Hospital of Brasilia, DF, Brazil
eCorresponding Author: Mirian Moura, SMHS 101 s/n Hospital Regional da Asa Norte, Brasilia, DF, CEP 70000-000, Brazil
Manuscript accepted for publication February 16, 2016
Short title: Stem Cell Therapy in ALS
doi: http://dx.doi.org/10.14740/jocmr2495w
| Abstract | ▴Top |
Background: Published studies seeking to improve survival in amyotrophic lateral sclerosis (ALS) have poor results in humans, although there are several studies in animal models with positive results.
Methods: We conducted a systematic review and meta-analysis of studies that were published between March 2009 and March 2015 on stem cell therapy and survival in animal models and patients with ALS. A total of 714 articles were identified, and from these, we selected preclinical in vivo studies and retrospective clinical studies.
Results and conclusions: A meta-analysis confirmed the efficacy of stem cell therapy in improving survival in preclinical trials, where a mean difference of 9.79 days (95% confidence interval: 4.45 - 15.14) in lifespan favored stem cell therapy. In contrast, the number of clinical studies is still insufficient to assess their effectiveness, and these studies only demonstrate the absence of serious adverse events. However, even this conclusion should be interpreted with caution because clinical studies are retrospective and heterogeneous and have an unsatisfactory quality.
Keywords: ALS; Amyotrophic lateral sclerosis; Motor neuron; Cell therapy; Survival
| Introduction | ▴Top |
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disease. It is frequently sporadic and characterized by the progressive degeneration of both upper and lower motor neurons in the brain, brainstem and spinal cord [1]. The incidence of ALS varies between 1.2 and 4.0 per 100,000 individuals per year, and ALS predominantly occurs in males [2, 3]. Death occurs between 2 and 4 years after onset due to respiratory insufficiency [1].
The mechanisms of ALS development are poorly understood, but the injury mechanisms of the disease may include both glial cells and neurons [4, 5]. The main known mechanisms of ALS pathogenesis are oxidative stress with damage to RNA, mitochondrial dysfunction, impairment of axonal transport, glutamate excitotoxicity, protein aggregation, endoplasmic reticulum stress, abnormal RNA processing, neuroinflammation and excitability of peripheral axons [6].
The clinical heterogeneity of ALS makes it difficult to identify the exact cause of the disease for the development of effective therapies. However, the drug riluzole may extend patient survival by a few months [7]. In addition, multidisciplinary care, enteral nutrition and non-invasive ventilation can extend patient survival [8].
The results of studies on the use of stem cell therapy to preserve motor neuron function have been considered without any evidence of their effectiveness in humans, although the use of this therapeutic approach has been fairly efficacious in experimental animal models [9].
Given the need to identify new alternatives to treat ALS, the aim of the present study was to conduct a systematic literature review to assess the efficacy of stem cell therapy in clinical and preclinical studies.
| Materials and Methods | ▴Top |
Strategies used to identify studies for meta-analysis
In May 2015, we investigated primary preclinical in vivo studies and clinical studies and subsequent meta-analyses published between March 2009 and March 2015 in the following electronic databases: Medline, Embase, Cochrane Library and Lilacs. The following Medical Subject Headings (MeSH) and Health Science Descriptors (HScDe) were used: “Amyotrophic Lateral Sclerosis” OR “Motor Neuron Disease” AND “Treatment” AND “cell therapy”.
Two authors independently evaluated the titles and abstracts of all studies identified in the search of the aforementioned electronic databases based on the descriptors.
The inclusion criteria were the following: 1) clinical, prospective or retrospective studies of patients diagnosed with a motor neuron disease by means of anamnesis and electromyography according to the El Escorial [10] and Awaji criteria [11]; 2) preclinical and in vivo studies with assessment of survival compared to a control group, and studies of treatment after the onset of weakness; and 3) studies based on the use of stem cell therapy to increase survival time compared to placebo or other treatments used by the control group.
The exclusion criteria were studies in which participants presented with respiratory failure or spinal muscular atrophy, studies in which the treatment was administered only prior to the disease onset, and narrative reviews, letters, editorials, case reports, duplicate publications or those without objective data to be evaluated.
Articles published in all languages were included.
The studies that met the inclusion criteria were obtained in full. References were also considered, and communication with the authors was established in cases of doubt. Disagreements were resolved by consensus, and when this was impossible, there was a subsequent analysis by two additional reviewers.
Data extraction
Data were obtained from each study using a review form with the following content: author, place where the work was conducted, year of publication, intervention, study design, number of participants, age, analysis by intention to treat, declaration of conflict of interest, evaluation by a research ethics committee, and animal species used if the study was preclinical. The following outcomes were assessed: 1) comparison between different treatments; 2) analysis of mean survival and absolute days of survival; 3) mean duration of the disease until the start of the intervention; 4) incidence of reactions and adverse effects of proposed treatments.
Assessing the quality of the studies
Quality was assessed by two independent authors, and in cases of disagreement, the situation was resolved by consensus among all authors. The Grading of Recommendations, Assessment, Development and Evaluation (GRADE) model [12] was used to access the quality of the studies. The following data were observed in the studies: 1) methods including research question, treatment sequence, allocation confidentiality, post-intervention follow-up, blinded outcome assessment, primary clinical outcome measures, location of study, protection against contamination, calculation of statistical power, sample representativeness, conflict of interest, and ethical aspects; 2) participants including inclusion criteria, exclusion criteria, age, gender, disease severity, and disease variants; 3) interventions including procedures, follow-up time, and method for monitoring disease progression; and 4) outcomes assessed in the review including disease duration before intervention and survival time.
The results of the primary outcome were obtained based on the intention-to-treat principle. For a continuous outcome, the following variables were calculated: mean, standard deviation and number of participants in each group. Data from work that was published more than once were obtained from the more thorough study.
The authors rated each primary study according to the overall quality of evidence as follows: A - high; B - moderate; C - low; and D - very low, assigning scores of 1 - 5 according to the number of biases.
For analytical purposes, the studies were grouped as 1) interventions in animal models and 2) clinical studies.
Statistical analysis
Statistical analysis was performed in preclinical trials using RevMan software, version 5.3. All P-values < 0.05 were considered to be statistically significant.
For continuous variables, such as animal survival in days, the weighted mean difference (random effects model) was calculated based on the DerSimonian and Laird method, with a corresponding 95% confidence interval (CI).
To evaluate the heterogeneity among the studies, a heterogeneity test was performed by calculating both the Q-test of heterogeneity and the I2 test of inconsistency. Heterogeneity was considered significant when P < 0.10. In addition, a sensitivity analysis was conducted using a funnel plot to quantify the presence of publication bias.
| Results | ▴Top |
Initially, 714 articles were obtained. Based on their abstracts, we selected one retrospective controlled clinical trial, 14 preclinical studies and 12 clinical uncontrolled descriptive studies.
After each original document was reviewed and data were obtained, four preclinical trials were excluded. Of these, one was an in vitro study [13], two did not include a quantitative evaluation of survival [14, 15], and one only reported survival ratios and proportions [16].
Finally, nine preclinical in vivo studies and 12 retrospective descriptive studies using stem cell therapy were included in the review. Although most of these studies were not controlled and randomized, they were included due to the limited number of studies that met this requirement. A flowchart illustrates the selection process that was adopted in the systematic review (Fig. 1).
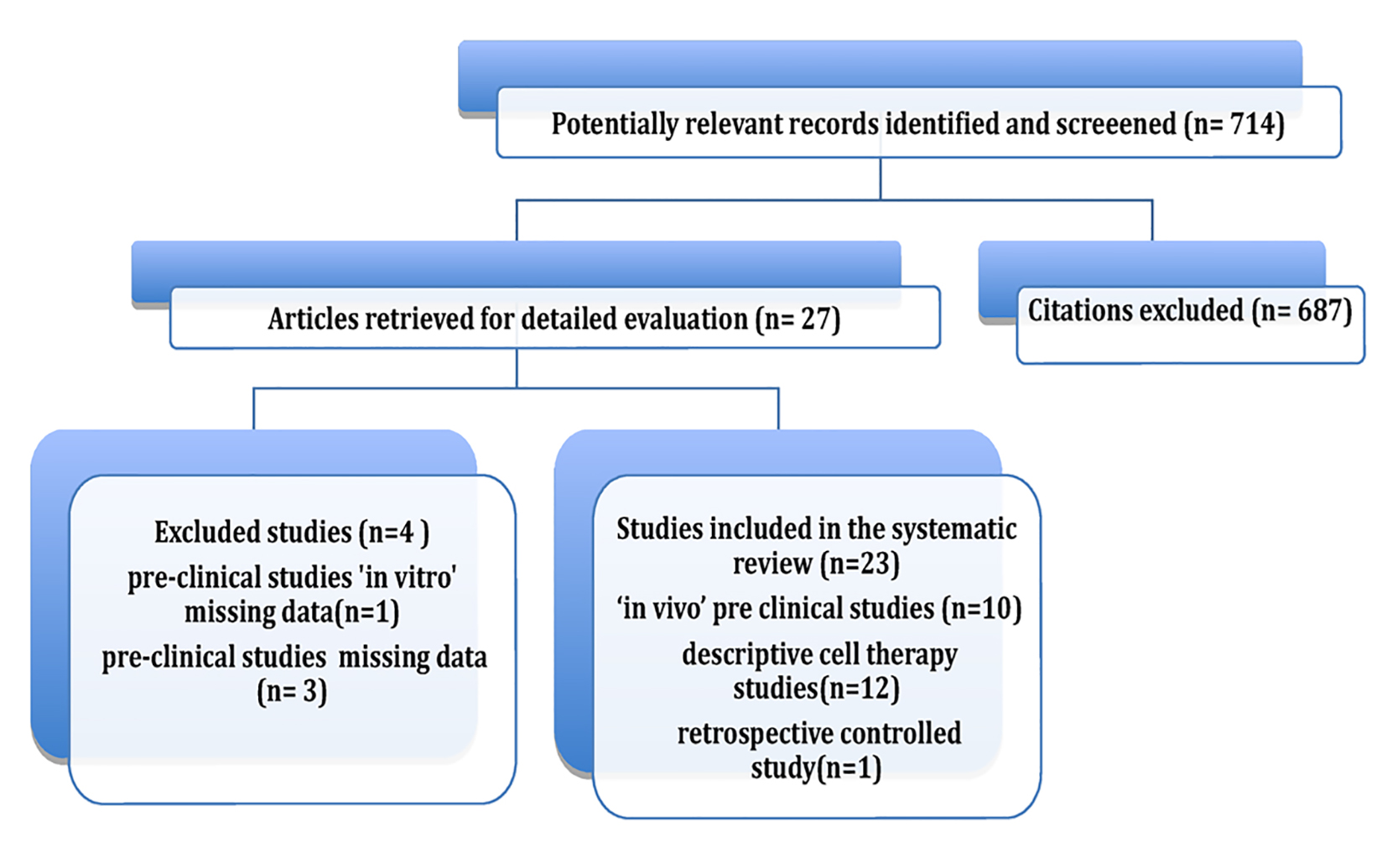 Click for large image | Figure 1. Review flowchart. |
Preclinical studies
Preclinical studies have been insightful for identifying the types of stem cells that offer therapeutic benefit in ALS. In vivo studies were conducted in transgenic mice expressing human mutated superoxide dismutase 1 and G93A(SOD1G93A) and presented experimental ALS treatments using neuroprotective therapies. These studies can be considered homogeneous with respect to their methods and their evaluation of outcomes in animals, which allows for a comparison using meta-analytical methods that involve a network meta-analysis [12]. All included studies analyzed survival using the Kaplan-Meier method with a log-rank test, but only those studies that clearly included the mean survival of animals in days and standard deviation were included in our meta-analysis. The authors of all of the studies that did not include such data were contacted via email.
Figure 2 shows an acyclic graph (forest) of cell therapy preclinical trials in animal models, demonstrating a mean meta-analytical difference of survival of 9.79 days (95% CI: 4.45 - 15.14), which favored cell therapy over placebo.
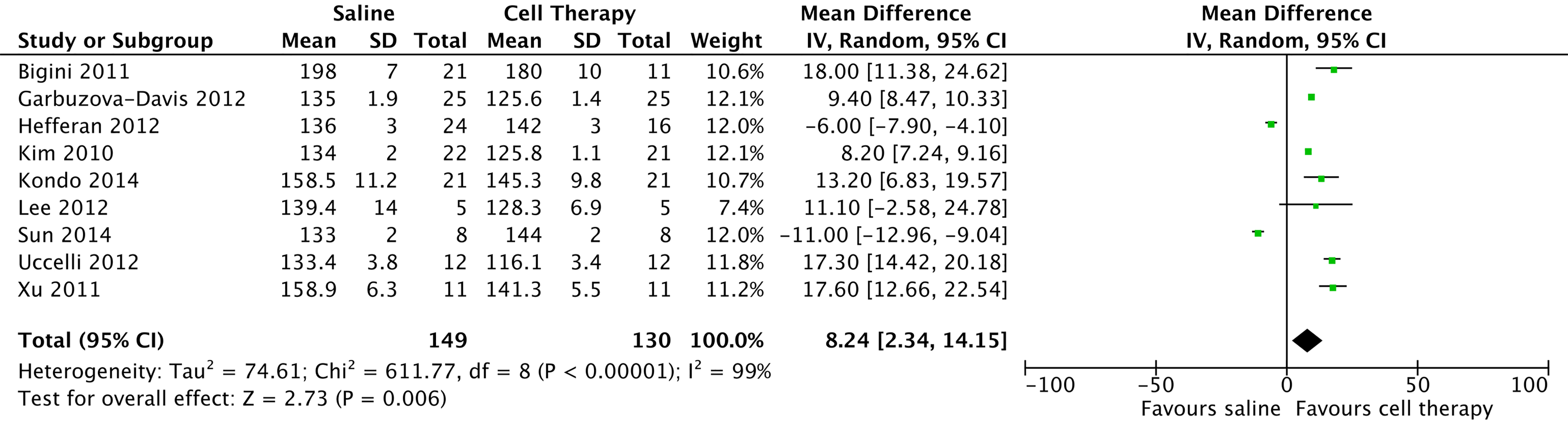 Click for large image | Figure 2. Preclinical studies in amyotrophic lateral sclerosis model with stem cell therapy, 2009 - 2015. |
In 2010, a study conducted in South Korea [17] showed that mesenchymal cell transplantation (ALS-hMSCs) at three different doses in association with cyclosporine was effective in slowing disease progression at high doses using an intrathecal injection of 1 × 106 cells/mL into the cisterna magna (P = 0.033) and an intravenous injection of neurotrophic factors. In 2011, in Italy [18], intraventricular administration of mononuclear cells from human umbilical cord showed that treated animals had a significantly longer survival than controls (P < 0.01, Gehan-Breslow-Wilcoxon test). A study in 2012 [19] showed that an intravenous injection of mesenchymal cells also led to increased survival of the animals (P < 0.005). In 2014, a study in Japan showed the efficacy of a transplantation of glial-rich neural progenitors in attenuating motor neuron degeneration and disease progression in rodent models [20].
In 2012, Garbuzova-Davis et al [21] performed weekly intravenous injections of cells from human umbilical cord in the pre-symptomatic stage and in the 13th week of the established disease stage. Two dosages were analyzed, 1 × 106 cells and 2.5 × 106 cells. The results showed that the greatest increases in survival occurred with the administration at the pre-symptomatic stage using the lowest concentration of cells (P = 0.0015) and at a dose of 2.5 × 106 for symptomatic animals (P = 0.0022).
Three studies examined the transplantation of stem cells from human fetuses into the anterior horn of the lumbar cord in immunosuppressed mice, before the onset of ALS symptoms [14, 22, 23]. In a study by Hefferan et al [22], improvement was transient and not very significant (P = 0.122), while a study by Xu et al [14] showed that a cervical and lumbar engraftment of cervical and thoracic spinal cord cells of an 8-week human fetus significantly prolonged survival by 17 days. The study was not analyzed in conjunction with the others because there were missing data.
In 2012, Lee et al [24] replaced microglia with a mutant SOD1 gene with microglia that expressed the wild-type gene using an injection of clodronate liposome and bone marrow transplants into the fourth cerebral ventricle and subsequently observed a 51.03% increase in survival.
In 2014, Sun et al reported the behavioral improvement and extended lifespan of ALS model mice transplanted with fetal human neural cells into the spinal cord. It was suggested that intrathecal transplantation of motor neurons into the lumbar spinal cord of animals migrated into the ventral horn area and improved ambulatory function and survival [25].
Also in 2014, Nizzardo et al [26] showed that survival in an animal model significantly benefited from both intrathecal and intravenous injections of specific neural stem cells that were derived from induced pluripotent stem cells. These positive effects are attributed to the activity of multiple mechanisms, including the production of neurotrophic factors and the reduction of microgliosis and macrogliosis.
In the studies described above, histopathological analyses of the nervous tissue of the spinal cord and brain were conducted after euthanasia of the animals, and a longer survival of motor neurons and glial cells was observed. Glial cells became branched after treatment but had a lower density and lower reactivity and showed less gliosis [18, 19, 21].
Clinical studies
Table 1 [27-38] summarizes the cell therapy clinical trials. These studies were conducted with the main objective of assessing the adverse effects inherent to the procedure and the post-procedure clinical evolution of the disease. Some of these effects were assessed using the ALSFRS-R. The mean disease duration was 2.32 ± 1.1 years and ranged from 0.7 to 4.4 years. The follow-up period ranged from 6 to 47.2 months. The number of patients per study ranged from 6 to 24 with an average of 11. All but one study were uncontrolled, with a quality of C or D.
 Click to view | Table 1. Studies in Amyotrophic Lateral Sclerosis Patients With Stem Cell Therapy, 2009 - 2015 |
In 2009, Martinez et al [27] performed a stereotactic autologous transplantation of CD133+ mononuclear cells into the frontal cortex, noting that the procedure is safe and well tolerated in patients with ALS. The survival time of the treated patients was statistically higher (P = 0.01) than that of the control group. In that same year, Mazzini et al [28] and Deda et al [29] injected autologous bone marrow cells into the anterior horn of the thoracic and cervical spinal cord, reporting pain and sensory loss as adverse effects; there was no significant functional decline compared to the control group, which confirmed the safety of the procedure.
An observational study conducted in Spain in 2010 [30] in which the use of cell therapy in 12 patients was evaluated found no beneficial effect. In that same year, two authors [31, 32] reported the effects of simultaneous intravenous and intrathecal space injections of autologous bone marrow cells [31]. One of them used granulocyte colony-stimulating factor (G-CSF) before the procedure [32] in a study of 24 patients. Although the study addressed issues of feasibility and safety, there were signs of clinical improvement that were associated with cell mobilization following the use of G-CSF. The authors conclude that prolonged monitoring and a greater number of patients are necessary.
Recently, three separate studies [33-35] showed that the use of cells derived from fetal spinal cord or umbilical cord for microinjection into the lumbar spine after laminectomy was associated with immunosuppression. There was hemorrhage in four patients without an associated functional deterioration; the adverse effects were attributed to immunosuppression. Mazzini et al used autologous mononuclear bone marrow cell transplantation with a 9-year follow-up and observed stabilization of FVC and ALSFRS-R scores [28]. However, the percentage of young individuals in that study who had a better prognosis (above 60%) has been reported as a confounding variable.
In a retrospective analytical study, Sharma et al [36] reported a better survival with the use of mononuclear stem cells; however, there were confounding factors in the experimental group, where the improvement of survival was also related to an age below 50 years and use of lithium. In this study, there were no important adverse events.
The remaining studies [30, 37, 38] also reported good tolerance to microinjections of fetal or autologous mesenchymal cells. In conclusion, only two studies [28, 36] showed improvement of survival, but with an unsatisfactory quality.
| Discussion | ▴Top |
Stem cell therapy is a promising potential treatment option for ALS, given the remarkable plasticity of stem cells and their ability to differentiate into multiple neuronal lineages. Stem cells can be used as important models for molecular pathway studies, drug screening, and cell therapy studies. Notably, there are two actual clinical trials, NeuralStem [39] and BrainStorm (NCT01051882).
This systematic review uses a meta-analysis to show the efficacy of stem cell therapy in improving survival in preclinical trials, whereas the number of clinical studies is still insufficient to assess their effectiveness and demonstrates only the absence of serious adverse events. However, caution is required because most clinical studies are heterogeneous with an unsatisfactory quality. Only one study was controlled but was retrospective and with confounding factors, such as the concurrent use of lithium and the interference of age in the analysis of survival.
To replace the cells that have undergone degeneration, various sources of stem cells can be used, such as bone marrow cells, neural stem cells, mesenchymal cells, astrocyte precursor cells and pluripotent cells [40]. Currently, there are basically two stem cell types for disease modeling, embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) [41].
The assessed preclinical trials tend to use relatively young mutant SOD1G93A mice in homogeneous groups in a controlled environment in which the animals showed a similar clinical condition. Although animal models are very useful for mimicking human diseases, they have limitations because they present with a distinct disease progression and show diverse responses in trials with drugs [3, 42]. Moreover, the sample size and sex of the animals often vary between studies.
According to some authors [6, 42], there are concerns about translating preclinical studies into effective human treatments. In preclinical studies, SOD1 animal models represented familial ALS more than sporadic ALS. In addition, ALS can be defined as a syndrome in which the pathophysiological mechanisms are poorly understood [4], and it is possible that familial and sporadic ALS differs in some fundamental mechanisms that determine the effectiveness of treatments.
Recent studies with ESCs have shown that the use of cell therapy to substitute motor neurons is not sufficient to impede the neurodegenerative process. In addition to the neuronal mechanisms, the toxic environment provided by glial cells contributes to motor neuron death. On the other hand, these findings have been limited to only one gene, SOD1, but the disease involves multiple pathways involving other genes such as C90ORF72, TDP-43, FUS and cytoplasmic aggregates, suggesting an underlying convergence of cellular processes [43, 44].
ESCs are found in the blastocyst and can differentiate into oligodendrocytes, astrocytes or neurons. Hematopoietic stem cells from the bone marrow (HSCs or MSCs) produce mesenchymal cells, as well as blood cells, which are also found in adipose tissue, umbilical cord, placenta and fetal tissues. According to Vercelli et al [45], mesenchymal cells can migrate to the spinal cord of mice, where they have neuroprotective actions, such as preventing the activation of microglia and the process of tissue gliosis and improving the count of motor neurons, which explains the positive results observed in all of the animal studies and the trend observed in human studies.
Another option for transplantation could be the use of stem cells that are derived from the olfactory epithelium (OECs). OECs continue to multiply during the postnatal period, are multipotent, and serve as conductive connections between the central and peripheral nervous systems [46]. In 2008, a clinical trial of 35 patients was conducted [47] and found that olfactory cell transplantation may slow disease progression. OEC transplantation for ALS has been performed in China with positive effects in spinal cord injury studies, such as axonal regeneration, remyelination and functional improvements. Although a large Chinese study reports that OECs may offer a benefit to ALS, other reports criticize the observed outcomes and do not support the clinical translation of this therapeutic approach at this time.
The histological analyses performed in all of these cell therapy studies show an improved animal survival, which supports the potential of this approach for neuroprotection with greater tissue preservation. However, among both cell therapy preclinical trials and cell therapy clinical trials, there were no studies with negative results, which may indicate a possible publication bias.
The development of iPSCs has led to remarkable changes in stem cell science. This technology has made it possible to obtain pluripotent stem cells directly from a patient’s adult cells. These cells are usually induced to form embryonic bodies and subsequently form neural precursor cells (NPCs) [41], which holds new promise for the treatment of neurodegenerative diseases.
However, studies have shown that there are many similarities between iPSCs and ESCs, such as telomere renewal during cell reprogramming into iPSCs and telomere shortening upon differentiation into somatic cells [48]. This similarity suggests that iPSCs could potentially be used as patient-specific ESCs, consequently preventing rejection and eliminating any ethical issues.
These recent studies in humans with ALS are summarized in Table 1 and include many differences, such as the number of patients, cell type, delivery method and outcome measurement strategies; however, each study has the potential to contribute to increasing our current understanding of the safety and feasibility of stem cell therapies for ALS. These studies were considered to be of low quality because of biases.
Moreover, in most studies, the cell therapy procedure was uncontrolled and performed in patients with a very advanced stage of disease. The disease onset was variable and frequently prolonged at 2.32 ± 1.1 years. Most authors agree that the treatment must be performed early in the course of the disease [41]. The goal of most of the studies was to assess adverse events and tolerability to treatment.
Guidelines have been introduced [49] that should reduce the number of false positives in preclinical studies and therefore prevent unnecessary clinical trials, which have occurred for various drugs. These recommendations include the following: 1) rigorously assessing an animal’s physical and biochemical characteristics with respect to human disease; 2) characterizing disease symptoms and the occurrence of death and being alert to unexpected variations; and 3) creating a mathematical model to address questions about the experimental design, such as the number mice that must be included in a study. To reduce concerns about animal research, Perrin [42] suggested excluding irrelevant animals, balancing for gender, avoiding the use siblings in the same treatment group, and tracking genes that induce non-inherited disease.
In conclusion, ALS is a rare heterogeneous disease that is still poorly understood in terms of its pathophysiology. Moreover, from a clinical point of view, ALS is difficult to manage. Preclinical studies of stem cell therapy show great efficacy, whereas more prospective and controlled studies are needed to establish the effectiveness of clinical studies in improving survival. Nonetheless, the most effective cell type to be used in transplantation must be determined, and it should be the one that shows better potential for neurogenesis and not only neuroprotective mechanisms.
Disclosures
The authors declare no conflicts of interest and have followed the sources of funding statement. The study was conducted by the Center of Reference for Neuromuscular Diseases, State Secretariat of Health of the Federal District, Brasilia, DF, Brazil.
Author Contributions
All authors participated in the design of the study, the analysis of the data, the interpretation of the results and the review of the manuscript; MCM, MRCGN, YSSPZ and EJE collected and analyzed data, and MCM, MRCGN and LAC wrote the manuscript and made final revisions.
| References | ▴Top |
- Gordon PH. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013;4(5):295-310.
doi pubmed - Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118-130.
doi pubmed - Marin B, Gil J, Preux PM, Funalot B, Couratier P. Incidence of amyotrophic lateral sclerosis in the Limousin region of France, 1997-2007. Amyotroph Lateral Scler. 2009;10(4):216-220.
doi pubmed - Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661-670.
doi pubmed - D'Amico E, Factor-Litvak P, Santella RM, Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic Biol Med. 2013;65:509-527.
doi pubmed - Mancuso R, Navarro X. Amyotrophic lateral sclerosis: Current perspectives from basic research to the clinic. Prog Neurobiol. 2015;133:1-26.
doi pubmed - Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;3:CD001447.
doi - Miller RG, Rosenberg JA, Gelinas DF, Mitsumoto H, Newman D, Sufit R, Borasio GD, et al. Practice parameter: The care of the patient with amyotrophic lateral sclerosis (An evidence-based review). Muscle Nerve. 1999;22(8):1104-1118.
doi - Lunn JS, Sakowski SA, Feldman EL. Concise review: Stem cell therapies for amyotrophic lateral sclerosis: recent advances and prospects for the future. Stem Cells. 2014;32(5):1099-1109.
doi pubmed - Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, Shefner J. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5-6):291-292.
doi pubmed - Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol. 2012;69(11):1410-1416.
doi pubmed - Puhan MA, Schunemann HJ, Murad MH, Li T, Brignardello-Petersen R, Singh JA, Kessels AG, et al. A GRADE Working Group approach for rating the quality of treatment effect estimates from network meta-analysis. BMJ. 2014;349:g5630.
doi pubmed - Gu R, Hou X, Pang R, Li L, Chen F, Geng J, Xu Y, et al. Human adipose-derived stem cells enhance the glutamate uptake function of GLT1 in SOD1(G93A)-bearing astrocytes. Biochem Biophys Res Commun. 2010;393(3):481-486.
doi pubmed - Xu L, Shen P, Hazel T, Johe K, Koliatsos VE. Dual transplantation of human neural stem cells into cervical and lumbar cord ameliorates motor neuron disease in SOD1 transgenic rats. Neurosci Lett. 2011;494(3):222-226.
doi pubmed - Boido M, Piras A, Valsecchi V, Spigolon G, Mareschi K, Ferrero I, Vizzini A, et al. Human mesenchymal stromal cell transplantation modulates neuroinflammatory milieu in a mouse model of amyotrophic lateral sclerosis. Cytotherapy. 2014;16(8):1059-1072.
doi pubmed - Dadon-Nachum M, Ben-Yaacov K, Ben-Zur T, Barhum Y, Yaffe D, Perlson E, Offen D. Transplanted modified muscle progenitor cells expressing a mixture of neurotrophic factors delay disease onset and enhance survival in the SOD1 mouse model of ALS. J Mol Neurosci. 2015;55(3):788-797.
doi pubmed - Kim H, Kim HY, Choi MR, Hwang S, Nam KH, Kim HC, Han JS, et al. Dose-dependent efficacy of ALS-human mesenchymal stem cells transplantation into cisterna magna in SOD1-G93A ALS mice. Neurosci Lett. 2010;468(3):190-194.
doi pubmed - Bigini P, Veglianese P, Andriolo G, Cova L, Grignaschi G, Caron I, Daleno C, et al. Intracerebroventricular administration of human umbilical cord blood cells delays disease progression in two murine models of motor neuron degeneration. Rejuvenation Res. 2011;14(6):623-639.
doi pubmed - Uccelli A, Milanese M, Principato MC, Morando S, Bonifacino T, Vergani L, Giunti D, et al. Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis. Mol Med. 2012;18:794-804.
doi pubmed - Kondo T, Funayama M, Tsukita K, Hotta A, Yasuda A, Nori S, Kaneko S, et al. Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Reports. 2014;3(2):242-249.
doi pubmed - Garbuzova-Davis S, Rodrigues MC, Mirtyl S, Turner S, Mitha S, Sodhi J, Suthakaran S, et al. Multiple intravenous administrations of human umbilical cord blood cells benefit in a mouse model of ALS. PLoS One. 2012;7(2):e31254.
doi pubmed - Hefferan MP, Galik J, Kakinohana O, Sekerkova G, Santucci C, Marsala S, Navarro R, et al. Human neural stem cell replacement therapy for amyotrophic lateral sclerosis by spinal transplantation. PLoS One. 2012;7(8):e42614.
doi pubmed - Lee HJ, Kim KS, Ahn J, Bae HM, Lim I, Kim SU. Human motor neurons generated from neural stem cells delay clinical onset and prolong life in ALS mouse model. PLoS One. 2014;9(5):e97518.
doi pubmed - Lee JC, Seong J, Kim SH, Lee SJ, Cho YJ, An J, Nam DH, et al. Replacement of microglial cells using Clodronate liposome and bone marrow transplantation in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2012;418(2):359-365.
doi pubmed - Sun H, Hou Z, Yang H, Meng M, Li P, Zou Q, Yang L, et al. Multiple systemic transplantations of human amniotic mesenchymal stem cells exert therapeutic effects in an ALS mouse model. Cell Tissue Res. 2014;357(3):571-582.
doi pubmed - Nizzardo M, Simone C, Rizzo F, Ruggieri M, Salani S, Riboldi G, Faravelli I, et al. Minimally invasive transplantation of iPSC-derived ALDHhiSSCloVLA4+ neural stem cells effectively improves the phenotype of an amyotrophic lateral sclerosis model. Hum Mol Genet. 2014;23(2):342-354.
doi pubmed - Martinez HR, Gonzalez-Garza MT, Moreno-Cuevas JE, Caro E, Gutierrez-Jimenez E, Segura JJ. Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy. 2009;11(1):26-34.
doi pubmed - Mazzini L, Mareschi K, Ferrero I, Miglioretti M, Stecco A, Servo S, Carriero A, et al. Mesenchymal stromal cell transplantation in amyotrophic lateral sclerosis: a long-term safety study. Cytotherapy. 2012;14(1):56-60.
doi pubmed - Deda H, Inci MC, Kurekci AE, Sav A, Kayihan K, Ozgun E, Ustunsoy GE, et al. Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-up. Cytotherapy. 2009;11(1):18-25.
doi pubmed - Gamez J, Carmona F, Raguer N, Ferrer-Sancho J, Martin-Henao GA, Marti-Beltran S, Badia M, et al. Cellular transplants in amyotrophic lateral sclerosis patients: an observational study. Cytotherapy. 2010;12(5):669-677.
doi pubmed - Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, Bulte JW, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67(10):1187-1194.
doi pubmed - Tarella C, Rutella S, Gualandi F, Melazzini M, Scime R, Petrini M, Moglia C, et al. Consistent bone marrow-derived cell mobilization following repeated short courses of granulocyte-colony-stimulating factor in patients with amyotrophic lateral sclerosis: results from a multicenter prospective trial. Cytotherapy. 2010;12(1):50-59.
doi pubmed - Glass JD, Boulis NM, Johe K, Rutkove SB, Federici T, Polak M, Kelly C, et al. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells. 2012;30(6):1144-1151.
doi pubmed - Riley J, Glass J, Feldman EL, Polak M, Bordeau J, Federici T, Johe K, et al. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I trial, cervical microinjection, and final surgical safety outcomes. Neurosurgery. 2014;74(1):77-87.
doi pubmed - Riley J, Federici T, Polak M, Kelly C, Glass J, Raore B, Taub J, et al. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I safety trial, technical note, and lumbar safety outcomes. Neurosurgery. 2012;71(2):405-416; discussion 416.
doi pubmed - Sharma AK, Sane HM, Paranjape AA, Gokulchandran N, Nagrajan A, D'sa M, Badhe PB. The effect of autologous bone marrow mononuclear cell transplantation on the survival duration in Amyotrophic Lateral Sclerosis - a retrospective controlled study. Am J Stem Cells. 2015;4(1):50-65.
pubmed - Blanquer M, Moraleda JM, Iniesta F, Gomez-Espuch J, Meca-Lallana J, Villaverde R, Perez-Espejo MA, et al. Neurotrophic bone marrow cellular nests prevent spinal motoneuron degeneration in amyotrophic lateral sclerosis patients: a pilot safety study. Stem Cells. 2012;30(6):1277-1285.
doi pubmed - Prabhakar S, Marwaha N, Lal V, Sharma RR, Rajan R, Khandelwal N. Autologous bone marrow-derived stem cells in amyotrophic lateral sclerosis: a pilot study. Neurol India. 2012;60(5):465-469.
doi pubmed - Feldman EL, Boulis NM, Hur J, Johe K, Rutkove SB, Federici T, Polak M, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol. 2014;75(3):363-373.
doi pubmed - McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D, Gottlieb DI, et al. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med. 1999;5(12):1410-1412.
doi pubmed - Coatti GC, Beccari MS, Olavio TR, Mitne-Neto M, Okamoto OK, Zatz M. Stem cells for amyotrophic lateral sclerosis modeling and therapy: myth or fact? Cytometry A. 2015;87(3):197-211.
doi pubmed - Perrin S. Preclinical research: Make mouse studies work. Nature. 2014;507(7493):423-425.
doi pubmed - Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 2011;122(6):657-671.
doi pubmed - Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(6):777-794.
doi pubmed - Vercelli A, Mereuta OM, Garbossa D, Muraca G, Mareschi K, Rustichelli D, Ferrero I, et al. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2008;31(3):395-405.
doi pubmed - Mackay-Sim A, St John JA. Olfactory ensheathing cells from the nose: clinical application in human spinal cord injuries. Exp Neurol. 2011;229(1):174-180.
doi pubmed - Huang H, Chen L, Xi H, Wang H, Zhang J, Zhang F, Liu Y. Fetal olfactory ensheathing cells transplantation in amyotrophic lateral sclerosis patients: a controlled pilot study. Clin Transplant. 2008;22(6):710-718.
doi pubmed - Gourronc FA, Klingelhutz AJ. Therapeutic opportunities: telomere maintenance in inducible pluripotent stem cells. Mutat Res. 2012;730(1-2):98-105.
doi pubmed - Ludolph AC, Bendotti C, Blaugrund E, Chio A, Greensmith L, Loeffler JP, Mead R, et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph Lateral Scler. 2010;11(1-2):38-45.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.