

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 8, Number 4, April 2016, pages 284-296
Signaling Pathways in Thyroid Cancer and Their Therapeutic Implications
Shan Jina, b, Oyungerel Borkhuua, Wuyuntu Baoa, Yun-Tian Yanga
aDepartment of General Surgery, Affiliated Hospital of Inner Mongolia Medical University, Hohhot 010050, Inner Mongolia Autonomous Region, China
bCorresponding Author: Shan Jin, Department of General Surgery, Affiliated Hospital of Inner Mongolia Medical University, Tongdao North Rd 1, Hohhot 010050, Inner Mongolia Autonomous Region, China
Manuscript accepted for publication February 11, 2016
Short title: Signaling Pathways in Thyroid Cancer
doi: http://dx.doi.org/10.14740/jocmr2480w
- Abstract
- Introduction
- Src Signaling Pathway
- Janus Kinase (JAK)-STAT Signaling Pathway
- MAPK Signaling Pathway
- PI3K/Akt Signaling Pathway
- NF-κB Signaling Pathway
- TSH Receptor (TSHR) Signaling Pathway
- Wnt-β-Catenin Signaling Pathway
- Notch Signaling Pathway
- Other Signaling Pathways
- References
| Abstract | ▴Top |
Thyroid cancer is a common malignancy of endocrine system, and has now become the fastest increasing cancer among all the malignancies. The development, progression, invasion, and metastasis are closely associated with multiple signaling pathways and the functions of related molecules, such as Src, Janus kinase (JAK)-signal transducers and activators of transcription (STAT), mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/Akt, NF-κB, thyroid stimulating hormone receptor (TSHR), Wnt-β-catenin and Notch signaling pathways. Each of the signaling pathways could exert its function singly or through network with other pathways. These pathways could cooperate, promote, antagonize, or interact with each other to form a complex network for the regulation. Dysfunction of this network could increase the development, progression, invasion, and metastasis of thyroid cancer. Inoperable thyroid cancer still has a poor prognosis. However, signaling pathway-related targeted therapies offer the hope of longer quality of meaningful life for this small group of patients. Signaling pathway-related targets provide unprecedented opportunities for further research and clinical development of novel treatment strategies for this cancer. In the present work, the advances in these signaling pathways and targeted treatments of thyroid cancer were reviewed.
Keywords: Thyroid cancer; Signaling pathway; Targeted therapy
| Introduction | ▴Top |
Thyroid cancer is a common malignancy of endocrine system, and its incidence rate is increasing year by year. In 2012, about 56,460 patients were newly diagnosed with thyroid cancer in the United States, resulting in 1,780 deaths. The incidence of thyroid cancer has increased by 4.99 folds from 1989 to 2012. About 62,980 new cases and 1,890 deaths were estimated in 2014 [1, 2]. Since 2002, the incidence rate of thyroid cancer in Korea has been increasing sharply with an annual increasing rate of 24.2% till 2010, and it has now become the most common type of cancer in the country [3]. In Beijing, about 1,099 cases of thyroid cancer were reportedly diagnosed in 2010, yielding an incidence rate of 8.78/100,000. Comparing to the incidence rate in 2001 (2.70/100,000), the incidence rate has increased by 225.2% in the past 9 years with an annual increasing rate of 14.2% [4]. Thyroid cancer has therefore become the most common cancer among all the malignancies. Several treatment modalities including surgical resection, radioactive iodide therapy, and hormone-suppressive therapy could result in good prognosis in most patients with differentiated thyroid cancer (DTC); however, such conventional treatment methods are not effective in treating patients with medullary thyroid cancer (MTC) or anaplastic thyroid cancer (ATC). About 2-5% of patients with DTC lose their differentiated features during treatment or natural course, making them difficult to get effectively treated by radioactive iodide treatment or thyroid stimulating hormone (TSH) suppressive therapy [5]. Distant metastases including pulmonary metastasis (50%), bone metastasis (25%), pulmonary and bone metastasis (20%), and other sites (5%) could be detected in about 10% of the patients and are the major causes of mortality [6]. Although conventional treatment methods are not effective for such patients, researches in molecular biology could bring new chances. The development, progression, invasion, and metastasis of thyroid cancer are closely associated with multiple signaling pathways and functions of related molecules. Any changes in these signaling pathways could be used as biomarkers to help diagnosis and predict the prognosis of thyroid cancer; in addition, potential treatment targets could also be found in these pathways. In the present work, the advances in these signaling pathways and targeted treatments of thyroid cancer were reviewed.
| Src Signaling Pathway | ▴Top |
Src family kinase (SFK) is a family of non-receptor tyrosine kinases. Nine members of SFKs have been identified till date including Src, Fyn, Yes, Blk, Fgr, Hck, Lck, Yrk, and Lyn. SFK could be activated by a variety of signals including tyrosine kinase, G protein-coupled receptors, steroid receptor, and signal transducers and activators of transcription (STAT). SFK could then be involved in cell proliferation, growth, motility, migration, angiogenesis, and intracellular transport [7, 8]. Both the direct and indirect activations of SFKs are associated with the progression and metastasis of malignancies. Previous studies have demonstrated that abnormal activation of SFKs is associated with leukemia, small cell lung cancer, non-small cell lung cancer, squamous cell carcinoma of the head and neck (SCCHN), breast cancer, prostate cancer, melanoma, and ovarian cancer. Drugs including dasatinib (BMS-354825), bosutinib (SKI-606), XL-228, KX01 (KX2-391), INNO-406 (NS-187), XL-999, and AZD-0530 which could target Src and SFKs have also been developed [8]. However, only very few studies have investigated the association between Src pathway and thyroid cancer. In an in vitro study, Henderson et al used Src inhibitors such as PP2, SU6656, and dasatinib, and the results showed that these inhibitors could effectively inhibit cell proliferation and expression of P-Src and P-FAK in papillary thyroid carcinoma (PTC) cell lines. Dasatinib could also significantly decrease tumor volume in mice carrying RET/PTC1 rearrangement, suggesting that Src pathway plays an important role in regulating PTC cell growth [9]. Another study also found that dasatinib could inhibit the growth of cancer cells, induce apoptosis and cell cycle arrest, and prevent tumor growth and metastasis, which all suggest that Src pathway is very important for the growth and metastasis of thyroid cancer, and Src inhibitors could effectively block thyroid cancer growth and metastasis [10]. SKI-606 could effectively reduce the tumor growth, invasion, and pulmonary metastasis of thyroid cancer in Thrb(PV/PV)Pten(+/-) mice, which could be caused by downregulating the Src pathway and inhibiting the epithelial-mesenchymal transition [11]. Similarly, ADZ0530 could effectively inhibit the cell growth and invasion of PTC and ATC by inhibiting Src-FAK pathway [12]. These findings suggest that the kinases of Src family play critical roles in the signal transduction of the development and progression of thyroid cancer. Activated Src pathway could consequently activate several other pathways including mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), FAK, and STAT pathways; these activations finally affect the growth, invasion, and metastasis of thyroid cancer [9-13].
| Janus Kinase (JAK)-STAT Signaling Pathway | ▴Top |
JAK is a family of non-receptor tyrosine kinases, which could be activated by the combination of cytokines or growth factors to the corresponding receptors. Activated JAK could in turn activate STAT [14, 15]. Four members of JAK family have been identified, which include JAK1, JAK2, JAK3, and TYK2. No individual correspondence exists between JAK and cytokines, which indicates that multiple JAKs could be activated by one cytokine, and several different cytokines could also activate one JAK [16]. STAT is a special family of proteins that could combine with deoxyribonucleic acid (DNA). Seven members of STAT family have been identified including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 [17]. After JAK induces phosphorylation, dimerization of STAT occurs to form STAT dimers, which could enter the cell nucleus and regulate the expression of target genes [18]. STAT3 has been acknowledged as a critical factor of JAK-STAT signaling pathway. Activated STAT3 could increase the expression of bcl-2 and surviving gene, which could in turn reduce the activity of caspase-3 and finally inhibit cell apoptosis [19, 20]. In addition, STAT3 could regulate the expression of genes that mediate survival genes (survivin, bcl-xl, mcl-1, and cellular FLICE-like inhibitory protein), proliferation genes (c-fos, c-myc, and cyclin D1), invasion genes (matrix metalloproteinase-2), and angiogenesis (vascular endothelial growth factor). STAT3 could be involved in the development and progression of tumors by collaborating with other factors including nuclear factor-κB (NF-κB) and hypoxia-inducible factor-1α (HIF1α) [21]. Studies demonstrated that STAT-3 inhibitors could reduce the tumor growth and induce cell apoptosis in SCCHN with activated STAT-3 [22]. Zhang et al [23] investigated STAT3 pathway in 49 patients with PTC (22 with and 27 without lymphatic metastases), and they found that the level of STAT3 expression in benign, non-neoplastic tissue is barely detectable. However, STAT3 expression was found in 11 of the 35 (31%) thyroid cancer tissues; pSTAT3 expression was found in only three of the 35 (9%) benign tissues, but in 40 of the 41 (98%) of the cancer tissues. In addition, pSTAT3 was detected in only four of the 22 (18%) lymph nodes from patients without lymphatic metastases, but in 12 of the 19 (59%) lymph nodes from patients with lymphatic metastases, among which, 45% were with strong staining, suggesting STAT3 pathway as a ubiquitous in PTC, which could activate pSTAT3 and promote the metastasis of PTC. Level of phospholipase D2 was found significantly higher in human PTC tissues than in normal tissues, which could collaborate with thyroid oncogenic kinase RET/PTC to activate STAT-3 [24]. High-fat diet was used to successfully induce ATC in Thrb(PV/PV)Pten(+/-) mice in a study by Kim et al, which suggested that high-fat diet induced the expression of two target genes of STAT3, namely, cyclin D1 and phosphorylated retinoblastoma protein encoding genes via JAK2-STAT3 pathway. STAT3 could thus promote the effects of obesity induced by high-fat diet on the development and progression of thyroid cancer in Thrb(PV/PV)Pten(+/-) mice [25]. These findings suggested the involvement of JAK-STAT3 pathway in the development, progression, and metastasis of thyroid cancer; however, Couto et al [26] found that the expression of tyrosine-phosphorylated or activated STAT3 (pY-STAT3) could be detected in patients with PTC (63/110, 57%), and the level of pY-STAT3 was negatively correlated with the size and distant metastasis of the tumors. STAT3 gene knock-out resulted in downregulation of multiple transcripts including tumor suppressor insulin-like growth factor binding protein 7; in addition, increase in glucose consumption, lactate production, and expression of HIF1α target genes were found after STAT3 gene knock-out, suggesting STAT3 as a negative regulator of aerobic glycolysis. Therefore, the researchers have suggested that STAT3 could also be a negative regulator of tumor growth [26], which is in accordance with the findings of Sosonkina et al [27]. Their research has also suggested that JAK/STAT3 pathway could only inhibit but not promote thyroid cancer [27]. In summary, the biological effects of JAK/STAT3 on thyroid cancer are very complicated; further studies are needed to clarify the associations between JAK/STAT3 pathway and thyroid cancer to help develop novel drugs for targeted therapy.
| MAPK Signaling Pathway | ▴Top |
MAPK is a family of intracellular serine/threonine protein kinases with four parallel MAPK signaling pathways identified in mammalian cells including extracellular-signal-regulated kinase (ERK), c-Jun N-terminal kinases (JNK/SAPK), p38MAPK, and ERK5/BMK1 [28, 29]. MAPK pathway could be activated by receptor tyrosine kinase, G protein-coupled receptors, and cytokines. It can then transduce extracellular signals into the cells and nuclei to induce biological responses including cell proliferation, differentiation, and apoptosis. Overactivation of MAPK pathway could upregulate the expression of numerous oncoproteins including chemokines, vascular endothelial growth factor A (VEGFA), matrix metalloproteinases (MMPs), prohibitin, vimentin, MET, NF-κB, HIF1α, EG-VEGF, transforming growth factor-β (TGFβ), and thrombospondin 1 (TSP1) [30]. Previous studies have demonstrated that increased activation of MAPK pathway, which is related with mutations of BRAF gene, is generally found in thyroid cancers [31, 32]. Nucera et al reported that 29-83% of thyroid cancer was accompanied by mutations of the BRAF gene [33]. Xing et al [34] reviewed 29 studies that investigated BRAFV600E variation in thyroid tumor and found that 44% (810/1,856) of PTC and 24% (23/94) of ATC involved BRAFV600E variation, while no BRAFV600E variation was found in the 165 follicular thyroid carcinomas (FTCs), 65 MTCs, and 542 benign thyroid tumors. These findings have suggested that BRAFV600E variation is closely associated with the development of PTC [34]. The findings that BRAFV600E variation is associated with pathological features including bigger PTC diameter, multifocality, extrathyroidal invasion, and lymph node metastasis [35, 36] have suggested BRAF mutation is closely associated with poor prognosis of PTC [37-39]. Vemurafenib, a selective BRAF inhibitor, could effectively be used in treating PTC with BRAFV600E mutation, suggesting that reduction in the activity of MAPK pathway by inhibiting BRAF is effective for treating thyroid cancer [40]. In another study, AZD6244, an MAPK pathway inhibitor, was administered to PTC cell line TPC1, and the proliferation rate of the cells decreased by 70%; treating TPC1 bearing nude mice with AZD6244 also increased the median time to progression to 32 days (the median time to progression was 10 days for the mice in the control group) [41]. In a multicenter clinical study, AZD6244 was used to treat 39 patients with 131I refractory PTC (IRPTC). After an evaluation based on the data of 32 patients, one partial response (3%), 21 stable disease (54%), and 11 progressive disease (28%) were found. Disease stability maintenance occurred at 16 weeks in 49% and 24 weeks in 36% of the patients, and the median progression-free survival (PFS) was 32 weeks. The median PFS of the patients with BRAFV600E variation (12/26, 46%) was 33 weeks, which was longer than that in the ones with wide type BRAF (11 weeks); however, the difference was not statistically significant (HR = 0.6, P = 0.3) [42]. MAPK pathway could also be regulated by upstream factors including receptor tyrosine kinases (RTKs), RAS oncogene, and RAF protein-serine/threonine kinases. Tyrosine kinase inhibitors (TKIs) including sorafenib [43-49], pazopanib [50, 51], axitinib [52], sunitinib [53-57], and motesanib [58, 59] could not only inhibit the MAPK pathway, but also inhibit several targets. TKIs have been considered as promising drugs for refractory thyroid cancers. Several ongoing phases I to III clinical trials with these inhibitors have already provided encouraging results. In November 2013, the United States Foods and Drugs Authority (FDA) approved the use of sorafenib for treating radioactive iodine-refractory differentiated thyroid cancer, which made sorafenib the third drug approved for targeted therapy of thyroid cancer (following vandetanib and cabozantinib) (Table 1 [40, 42-93]).
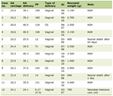 Click to view | Table 1. Drugs Targeting the Signaling Pathways in Thyroid Cancer Studied in Current Clinical Trials |
| PI3K/Akt Signaling Pathway | ▴Top |
PI3K is a complex present in cytoplasm, which could catalyze the phosphorylation of D3 position of phosphatidylinositol. Protein kinase B (PKB), a protein encoded by retroviral oncogene υ-Akt, is a downstream target protein of PI3K. Phosphorylated Akt (p-Akt), which is the activated form of Akt, could enter cytoplasm or cellular nuclei to phosphorylate a series of substrates and therefore exert biological functions. PI3K/Akt pathway is a very important intracellular signaling pathway for mammals, which is closely associated with cell proliferation, transformation, metabolism, motility, and development and progression of tumors [94]. Activated PI3K/Akt pathway could also activate other pathways including Wnt-β-catenin, HIF1α, FOXO3, and NF-κB pathways [95-97]. PI3K-activated Akt could in turn phosphorylate a series of downstream target proteins including Bad, caspase 9, forkhead, Par-4, p2l, and mammalian target of rapamycin (mTOR) to activate or inhibit their functions, which finally promote cell survival. Therefore, Akt has been regarded as an antiapoptotic regulator [98]. mTOR is also a member of PI3K protein kinase family and an important downstream molecular of PI3K pathway. PI3K/Akt/mTOR pathway could induce the development of tumor via various pathways; for instance, activation of mTOR could inhibit autophagy as a critical regulator in the initial period of autophagy. Activated Akt could inhibit the activation of caspase 3 and caspase 9 by phosphorylate Ser 196 and thus inhibit apoptosis; PI3K/Akt pathway could inactivate Bax by phosphorylate Ser 184 to inhibit apoptosis, and PI3K/Akt could induce phosphorylation of FOXO-1, which could enter cellular nuclei and expression to regulate cell cycle [99-102]. PI3K/Akt pathway could also promote tumor metastasis by promoting cell motility and angiogenesis; for instance, activated Akt could increase the activity of NF-κB and thus increase the motility of tumor cells. Activation of p70s6k, a protein downstream of mTOR, could increase cell motility. PI3K/Akt pathway could upregulate the expression of MMP-2 mRNA and protein, which could degrade extracellular matrix and promote tumor metastasis and invasion. PI3K/Akt could upregulate the expression of HIF1α and induce the transcription of VEGF gene to increase the expression of VEGF, which in turn induce angiogenesis and increase blood supply of tumors [103, 104]. RAS mutation is the second most gene mutation in thyroid cancer, which mainly activates PI3K/Akt pathway [105, 106]. Studies have reported that about 93% of FTCs and 96% of ATCs have mutations of the genes that encode proteins involved in PI3K/Akt pathway. These mutations include mutations of RTK, RAS, and phosphatase and tensin homolog deleted on chromosome 10 (PTEN) encoding genes, extra copies of phosphoinositide-3 kinase catalytic α (PIK3C A), phosphoinositide-3 kinase catalytic β (PIK3C B), 3-phosphoinositide-dependent kinase-1 (PDK1) encoding genes, and rearrangement of peroxisome proliferators-activated receptor γ/paired box gene 8 (PPARγ/Paχ8) encoding gene [107]. Xing et al suggested that activation of MAPK pathway could increase the development of PTC, which could further progress to ATC after dedifferentiation induced by activated PI3K/Akt pathway; in addition, PI3K/Akt pathway activation could induce the development of FTC, which could also progress to ATC after the induction caused by further activated PI3K/Akt pathway and/or MAPK pathway [108]. Administration of rapamycin (an mTOR inhibitor) to PTC1 could reduce the proliferation rate of the cells by 80%; animal study also demonstrated that rapamycin could increase the median time to progression to 23 days (the median time to progression was 10 days for control group) in TPC1 bearing nude mice [41]. Similarly, GDC-0941, an inhibitor of PI3K/Akt pathway could inhibit the compensatory survival response of thyroid cancer cells by arresting the cells at G1 phase and finally increase cell apoptosis [109]. Except for PI3K pathway, GDC-0941 could also inhibit HIF-1α pathway to reduce the metastasis of thyroid cancer [110]. Currently, many drugs including temsirolimus, everolimus, rapamycin, INK-128, OSI027, AZD8055, GSK690693, MK-2206, CAL-101, BYL719, PX866, GDC0941, BKM120, and ZSTK474 that target PI3K/Akt have been investigated in phases I to III clinical trials [111], among which, temsirolimus and everolimus have been found with remarkable efficacies in treating thyroid cancers [61-63].
| NF-κB Signaling Pathway | ▴Top |
NF-κB is a family of proteins that are homo- or heterodimers of Rel family members. Five NF-κB/Rel family members including Rel (cRel), p65 (RelA, NF-κB3), RelB, p50 (NF-κB1), and p52 (NF-κB2) have been identified in mammalian cells. IκB, another family of protein consisting of IKKα, IKKβ, IKKγ, IKKδ, IKKε, and BCL-3 that could inhibit the activity of NF-κB, has also been identified in cytoplasm. IκB mainly combines with NF-κB/Rel proteins in cytoplasm to keep them sequestered. Extracellular signals could activate IκB kinase (IKK) to phosphorylate IκB and degrade IκBα; the released p50/p65 dimer then enters the cell nuclei to combine with special sequence of κB and in turn induce the transcription of target genes [112, 113]. The association between NF-κB and development of tumor mainly relies on the fact that NF-κB could inhibit cell apoptosis. For instance, NF-κB could influence cell apoptosis by regulating cytokines including TNF-α, IL-1β, IL-6, and IL-8; it could also inhibit apoptosis by inducing or upregulating the expression of antiapoptotic gene such as bcl-2 or by inducing the expression of TNF-α receptor family (TRAF1 and TRAF2), cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2), and zinc finger protein A20 [114, 115]. Previous studies have demonstrated that NF-κB pathway is activated in several malignancies including leukemia, breast cancer, pancreas cancer, and thyroid cancer [116-120]. Specifically for thyroid cancer, NF-κB activation was found in PTCs, FTCs, and ATCs, and researchers suggested that activation of NF-κB could promote dedifferentiation of PTCs and FTCs and thus play important roles in each stage of thyroid cancer [119-122]. Activation of NF-κB could upregulate the expression of COX-2, IL-8, and GST-π in FTCs [120]. BRAFV600E could increase the carcinogenicity of thyroid cancer cells through NF-κB mediated c-IAP1, c-IAP2, and XIAP, thus increasing the invasiveness of thyroid cancer cells [123, 124]. Yamashita et al [125] suggested that mutations of BRAF and RAS genes and rearrangement of RET/PET gene could activate MAPK pathway, which in turn activate NF-κB pathway and increase the progression and invasiveness of PTCs. NF-κB could regulate the expression of MMPs, urokinase-type plasminogen activator (uPA), and IL-8 to allow cancer cells obtain invasiveness, which could then infiltrate surrounding tissues and metastasize to distant organs. Specifically, MMPs could dissolve and damage extracellular matrix and thus increase the infiltration and metastasis of cancer cells. Komorowski et al demonstrated that level of MMP-2 was significantly increased in PTC, and levels of MMP-3 and MMP-9 were significantly increased in MTC, suggesting that NF-κB activation could increase the invasiveness and metastasis of thyroid cancer cells [126]. Inhibition of NF-κB activation could affect the growth, apoptosis, and invasion of thyroid cancer cells [127]. In addition, activation of NF-κB could be inhibited when the activities of IKKs and proteasome are inhibited, or p50/p65 dimers are prevented from entering cell nuclei or combining with target DNA [112, 113]. Small-molecule triptolide, an NF-κB inhibitor, was used to treat two human ATC cell lines, namely TA-K cells and 8505C cells. The expression of cyclinD1, VEGF, and uPA was effectively inhibited, which in turn effectively reduced the angiogenesis and invasion of ATC [122]. Bortezomib, an inhibitor of proteasome, could increase the expression of p21(CIP1/WAF1) and thus inhibit cell growth, increase cell apoptosis, and arrest cells at G2-M phase in two ATC cell lines, namely C643 and SW1736 cells [128]. Currently, bortezomib has been approved by FDA to treat myeloma; however, more clinical studies are needed to evaluate the efficacy and safety of treating thyroid cancer with bortezomib [64].
| TSH Receptor (TSHR) Signaling Pathway | ▴Top |
TSHR is a guanine nucleotide-binding G protein-coupled receptor, which could combine with TSH and thus stimulate the growth of thyrocytes directly or indirectly by stimulating growth factors including autocrine growth factors and VEGF; in addition, sodium iodide symporter (NIS) promoter and upstream enhancers could also be activated and thus upregulate the expression of NIS, which could in turn increase the transport of iodide to cellular membrane [129]. Two intracellular signaling pathways, namely Gsα-mediated adenylyl cyclase-cyclic AMP (cAMP) signaling pathway and Gq- or G11-mediatedphospholipase C β-inositol 1,4,5-trisphosphate-intracellular Ca2+ signaling pathway, could be activated after the combination of TSHR with TSH [7]. Each signaling molecule in these pathways could interact with the signaling molecules of other pathways including Wnt, PI3K, and MAPK pathways to form a network [130]. Liu et al investigated 34 paraffin-embedding specimens of classical PTCs and 39 PTCs with tall-cell features, and they found that the positive expression rate of TSHR was significantly lower in PTCs than in the normal thyroid tissues adjacent to the cancers (χ2 = 15.70, P < 0.05) and lower in PTCs with tall-cell features than in classical PTCs (χ2 = 4.24, P < 0.05) [131]. Other studies also demonstrated that mRNA levels of NIS and TSHR were significantly lower in thyroid cancer tissues than in benign thyroid nodules or normal thyroid tissues [132]. TSHR expression could be very low in DTC and even undetectable in poorly differentiated carcinoma of the thyroid [133, 134]. During treatment or natural course, TSHR expression and iodide uptake could decrease in about 2-5% of DTCs, degenerative changes of the morphology and functions of thyroid cancer cells could also occur, and the cells could then lose their differentiated features and become insensitive to radioiodine therapy and TSH suppressive therapy [5]. Xing et al [135] suggested that decrease or lack of TSHR expression in human thyroid cancer tissues or thyroid cancer cell lines could be caused by the methylation of the gene promoter. DNA methylation is an epigenetic change which mainly occurs at the cytosine at or near the promoter of genes, which could regulate the transcription of these genes and cause gene silence, and it could thus decrease or totally inhibit the expression of corresponding protein [136]. Till date, over 25 mutations have been identified on TSHR gene [137, 138]. Clinical and in vitro studies have demonstrated that retinoic acid could induce redifferentiation of thyroid cancer cells, increase the expression of NIS and uptake of radioactive iodine, and therefore increase the efficacy of radioiodine therapy [139, 140]. Histone deacetylase inhibitor could increase the expression of NIS gene through posttranscriptional modification. Triehostatin A (TSA), a histone deacetylase inhibitor, could significantly increase the expression of NIS, induce the redifferentiation, and restore 131I uptake in three thyroid cancer cell lines (TPC-1, XTC-1, and FTC-133) [141]. Single or combined use of RDEA119 (a MAPK pathway inhibitor) and perifosine (a PI3K/Akt pathway inhibitor) could increase the expression of iodide-handling genes in 11 thyroid cancer cell lines. Additional use of SAHA (a histone deacetylase inhibitor) could increase the expression of NIS, TPO, and TSHR genes by about 2,500, 1,000, and 600 folds, respectively. 125I uptake was increased by three, five, and four folds for K1, C643, and KAT18 cells, respectively [142]. Sunitinib could also increase the expression of NIS gene by inhibiting MEK/ERK and SAPK/JNK pathways in PTCs [143]. These findings demonstrated that TSHR could collaborate with other pathways to form a network, and inhibition of TSHR pathway could increase the activity of other pathways, which could increase the growth of cancer cells, decrease the expression of NIS, and thus reduce iodide uptake of cancer cells. Therefore, targeting TSHR is an optimal method to induce the redifferentiation of poorly differentiated carcinoma of the thyroid and increase the iodide uptake of iodine-refractory thyroid carcinomas by increasing the NIS expression [144].
| Wnt-β-Catenin Signaling Pathway | ▴Top |
Wnt pathway is named after Wnt protein, a protein that initiates this pathway. It is associated with cell development and differentiation. Wnt pathway includes canonical Wnt pathway (Wnt-β-catenin pathway) and non-canonical pathway (Wnt-Ca2+ or Wnt-NK pathway) [145]. When activated, Wnt could combine with its receptor Fzd to activate Dsh protein, which is located inside the cells. Phosphorylated Dsh protein could then transduce the signals into the cells to inhibit the activity of the complex made up of APC, GSK-3β, Axin, and β-catenin, which could cause the accumulation of β-catenin in the cells. β-catenin could enter the cell nuclei and combine with transcription factors of Tcf/LEF family to form a complex, which could activate the transcription of downstream target genes including c-myc and cyclin D1 to increase the differentiation and proliferation of tumor cells [146]. Kurihara et al investigated the associations between Wnt-β-catenin pathway and ATC, and they found that β-catenin expression could be detected in 40.9% of the cell nuclei and 63.6% of the cytoplasm for the 22 patients with ATC; mutation rate of β-catenin, APC, and Axin1 genes were 4.5%, 9.0%, and 81.8%, respectively. The expressions of cyclin D1 and c-myc, two targets of Wnt-β-catenin pathway, were as high as 27.3% and 59.1%, respectively, suggesting that changes of Wnt-β-catenin pathway could be associated with the development of ATC [147]. Wnt-β-catenin could also regulate the expression of cyclin D1, which is associated with lymph node metastases in PTCs [148]. Interactions between β-catenin and E-cadherin could affect cell adhesion. Studies showed that E-cadherin could combine with cytoplasmic β-catenin to form E-cadherin/β-catenin/α-catenin complex, which in turn combines to cortical actin cytoskeleton to maintain cell stability and polarity of during adhesion that is essential for the integrity and function of epithelial tissues. Downregulation or absence of cell adhesion molecules including E-cadherin could induce the changes from keratin-dominant cytoskeleton to vimentin-dominant cytoskeleton, which could change the cell morphology, increase the motility of the cells, and thus increase the risk of tumor metastasis. Overexpression of E-cadherin could block the transcription ability of β-catenin, effectively shut-down the expression of target genes, and thus inhibit the proliferation and migration of the cells [149]. Dickkopf-1, a Wnt-β-catenin pathway inhibitor, was used to treat human PTC cell lines (including SNU-790, B-CPAP, and BHP10-3). Dickkopf-1 could effectively inhibit the survival and migration of PTC cells by regulating Wnt-β-catenin pathway and E-cadherin expression [150]. Antisense oligonucleotides, RNA interference, and neutralizing antibodies could be used for the targeted inhibition of Wnt-β-catenin pathway [151]. Rao et al treated ATC cell lines with imatinib mesylate, a specific tyrosine kinase inhibitor, and they found that imatinib mesylate could reduce the expression of β-catenin, stabilize the β-catenin/E-cadherin complex, and decrease the invasiveness of thyroid cancer; therefore, imatinib mesylate could be used as a drug for targeted therapy for some patients with ATC [152].
| Notch Signaling Pathway | ▴Top |
Notch pathway is composed of receptor, ligand, and intracellular effector CSL (CBF1/RBP-Jκ, Su(H), Lag1). Combination of Notch ligand and receptor could trigger two proteolyses. As a result of proteolysis, the Notch receptor then enters the nuclei and combines with downstream CSL to form a transcriptional activation complex and initiate transcription of target genes. Notch pathway could not only inhibit the differentiation of T cells and granulocyte, neurogenesis, and muscle formation, but also directly induce the ordered cell differentiation [153, 154]. Dysfunction of Notch pathway is associated with several biological processes including cellular function, microenvironment, cell proliferation, apoptosis, adhesion, epithelial-mesenchymal transition, and angiogenesis that are related to development of numerous tumors. Except for directly inducing the development of tumor, Notch pathway could also interact with many other pathways and thus indirectly induce the development of tumors [155]. Cancer treatments that target Notch pathway include γ-secretase inhibitors (GSIs), antibodies to Notch receptors, and Notch-1 siRNA. Previous clinical trials have demonstrated that monoclonal antibodies have shown efficacy against metastatic thyroid cancer, non-small cell lung cancer, sarcoma, colorectal cancer, melanoma, and ovarian cancer with GSIs [156]. Notch signal could be expressed in normal thyroid tissues and adenomas but not in ATCs. Overexpression of Notch signal could induce redifferentiation of thyroid cancer cells, which could reduce the growth of the cancer cells and directly affect NIS promoter to increase the expression NIS. This suggests that Notch pathway plays a role in the development, progression, and differentiation of thyroid cancer [157]. Activation of Notch pathway could effectively inhibit the cell growth of ATC in vivo and in vitro, and it inhibited the growth of MTC in vivo [158, 159]. Valproic acid was used to activate Notch 1 signal to induce apoptosis and inhibit cell growth in MTC cells, which provided experimental evidence for using such drugs in treating advanced MTCs [160]. Patel et al treated ATC cell line HTh7 with hesperetin, a Notch pathway activator, and they found that hesperetin could induce the expression of TTF1, TTF2, paired box gene 8, TSHR, and NIS; it thus improved the redifferentiation of ATC cells and inhibited ATC cell proliferation by inducing apoptosis [161]. In summary, Notch signaling pathway is closely associated with the development of thyroid cancer, and it could be an important target for treating thyroid cancer.
| Other Signaling Pathways | ▴Top |
Some other signaling pathways are also associated with the development and progression of thyroid cancer. For instance, RASSF1-MST1-FOXO3 pathway is associated with BRAFV600E gene mutation [162], C-met pathway is associated with the growth, invasion, and lymph node metastasis of thyroid cancer [163, 164], and sonic hedgehog pathway is ubiquitously expressed in thyroid cancers and is associated with the development, staging, and lymph node metastasis of thyroid cancer [165].
In summary, each of the signaling pathways could exert its function singly or through network with other pathways. These pathways could cooperate, promote, antagonize, or interact with each other to form a complex network for the regulation. Dysfunction of this network could increase the development, progression, invasion, and metastasis of thyroid cancer. Signaling pathway-related targeted therapy could be the fourth treatment method following surgical removal, radioiodine therapy, and endocrine suppressive therapy. In addition, it could induce the redifferentiation of residual thyroid cancer to achieve “benign” change, and performing immune induction to improve the ability against cancer is also very important in treating thyroid cancer.
Grant Support
This work is supported by the National Natural Science Foundation of China (Grant No. 81460157) and Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (Grant No. NJYT-14-B18).
Conflict of Interest
None.
| References | ▴Top |
- Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220-241.
doi pubmed - Pellegriti G, Frasca F, Regalbuto C, Squatrito S, Vigneri R. Worldwide increasing incidence of thyroid cancer: update on epidemiology and risk factors. J Cancer Epidemiol. 2013;2013:965212.
doi pubmed - Jung KW, Won YJ, Kong HJ, Oh CM, Seo HG, Lee JS. Cancer statistics in Korea: incidence, mortality, survival and prevalence in 2010. Cancer Res Treat. 2013;45(1):1-14.
doi pubmed - Peng JH. Recent cancer statistics in China. China Medical Tribune 2013-3-7. http://cancer.cmt.com.cn/detail/168613.html.
- Rivera M, Ghossein RA, Schoder H, Gomez D, Larson SM, Tuttle RM. Histopathologic characterization of radioactive iodine-refractory fluorodeoxyglucose-positron emission tomography-positive thyroid carcinoma. Cancer. 2008;113(1):48-56.
doi pubmed - Saez JM. Treatment directed to signalling molecules in patients with advanced differentiated thyroid cancer. Anticancer Agents Med Chem. 2013;13(3):483-495.
pubmed - Dempke W, Zippel R. [SRC kinases in tumor therapy]. Med Klin (Munich). 2010;105(10):711-715.
doi pubmed - Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14(7):667-678.
doi pubmed - Henderson YC, Toro-Serra R, Chen Y, Ryu J, Frederick MJ, Zhou G, Gallick GE, et al. Src inhibitors in suppression of papillary thyroid carcinoma growth. Head Neck. 2014;36(3):375-384.
doi pubmed - Chan CM, Jing X, Pike LA, Zhou Q, Lim DJ, Sams SB, Lund GS, et al. Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin Cancer Res. 2012;18(13):3580-3591.
doi pubmed - Kim WG, Guigon CJ, Fozzatti L, Park JW, Lu C, Willingham MC, Cheng SY. SKI-606, an Src inhibitor, reduces tumor growth, invasion, and distant metastasis in a mouse model of thyroid cancer. Clin Cancer Res. 2012;18(5):1281-1290.
doi pubmed - Schweppe RE, Kerege AA, French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009;94(6):2199-2203.
doi pubmed - Plaza Menacho I, Koster R, van der Sloot AM, Quax WJ, Osinga J, van der Sluis T, Hollema H, et al. RET-familial medullary thyroid carcinoma mutants Y791F and S891A activate a Src/JAK/STAT3 pathway, independent of glial cell line-derived neurotrophic factor. Cancer Res. 2005;65(5):1729-1737.
doi pubmed - Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18(11):545-551.
doi pubmed - Proia DA, Foley KP, Korbut T, Sang J, Smith D, Bates RC, Liu Y, et al. Multifaceted intervention by the Hsp90 inhibitor ganetespib (STA-9090) in cancer cells with activated JAK/STAT signaling. PLoS One. 2011;6(4):e18552.
doi pubmed - Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228(1):273-287.
doi pubmed - Mitchell TJ, John S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology. 2005;114(3):301-312.
doi pubmed - Wang YH, Huang ML. Organogenesis and tumorigenesis: insight from the JAK/STAT pathway in the Drosophila eye. Dev Dyn. 2010;239(10):2522-2533.
doi pubmed - Guerriero ML, Dudka A, Underhill-Day N, Heath JK, Priami C. Narrative-based computational modelling of the Gp130/JAK/STAT signalling pathway. BMC Syst Biol. 2009;3:40.
doi pubmed - Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol. 2008;19(4):311-318.
doi pubmed - Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59-76.
doi pubmed - Shim SH, Sung MW, Park SW, Heo DS. Absence of STAT1 disturbs the anticancer effect induced by STAT3 inhibition in head and neck carcinoma cell lines. Int J Mol Med. 2009;23(6):805-810.
pubmed - Zhang J, Gill A, Atmore B, Johns A, Delbridge L, Lai R, McMullen T. Upregulation of the signal transducers and activators of transcription 3 (STAT3) pathway in lymphatic metastases of papillary thyroid cancer. Int J Clin Exp Pathol. 2011;4(4):356-362.
pubmed - Kim YR, Byun HS, Won M, Park KA, Kim JM, Choi BL, Lee H, et al. Modulatory role of phospholipase D in the activation of signal transducer and activator of transcription (STAT)-3 by thyroid oncogenic kinase RET/PTC. BMC Cancer. 2008;8:144.
doi pubmed - Kim WG, Park JW, Willingham MC, Cheng SY. Diet-induced obesity increases tumor growth and promotes anaplastic change in thyroid cancer in a mouse model. Endocrinology. 2013;154(8):2936-2947.
doi pubmed - Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simoes M, Lima J, et al. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci U S A. 2012;109(35):E2361-2370.
doi pubmed - Sosonkina N, Starenki D, Park JI. The Role of STAT3 in Thyroid Cancer. Cancers (Basel). 2014;6(1):526-544.
doi pubmed - Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16(7):793-798.
doi pubmed - Akasaka E, Takekoshi S, Horikoshi Y, Toriumi K, Ikoma N, Mabuchi T, Tamiya S, et al. Protein oxidative damage and heme oxygenase in sunlight-exposed human skin: roles of MAPK responses to oxidative stress. Tokai J Exp Clin Med. 2010;35(4):152-164.
pubmed - Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13(3):184-199.
doi pubmed - Vidal AP, Andrade BM, Vaisman F, Cazarin J, Pinto LF, Breitenbach MM, Corbo R, et al. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur J Endocrinol. 2013;169(4):521-528.
doi pubmed - Leonardi GC, Candido S, Carbone M, Raiti F, Colaianni V, Garozzo S, Cina D, et al. BRAF mutations in papillary thyroid carcinoma and emerging targeted therapies (review). Mol Med Rep. 2012;6(4):687-694.
pubmed - Nucera C, Lawler J, Parangi S. BRAF(V600E) and microenvironment in thyroid cancer: a functional link to drive cancer progression. Cancer Res. 2011;71(7):2417-2422.
doi pubmed - Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28(7):742-762.
doi pubmed - Lim JY, Hong SW, Lee YS, Kim BW, Park CS, Chang HS, Cho JY. Clinicopathologic implications of the BRAF(V600E) mutation in papillary thyroid cancer: a subgroup analysis of 3130 cases in a single center. Thyroid. 2013;23(11):1423-1430.
doi pubmed - Li C, Lee KC, Schneider EB, Zeiger MA. BRAF V600E mutation and its association with clinicopathological features of papillary thyroid cancer: a meta-analysis. J Clin Endocrinol Metab. 2012;97(12):4559-4570.
doi pubmed - Elisei R, Viola D, Torregrossa L, Giannini R, Romei C, Ugolini C, Molinaro E, et al. The BRAF(V600E) mutation is an independent, poor prognostic factor for the outcome of patients with low-risk intrathyroid papillary thyroid carcinoma: single-institution results from a large cohort study. J Clin Endocrinol Metab. 2012;97(12):4390-4398.
doi pubmed - Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, Yip L, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA. 2013;309(14):1493-1501.
doi pubmed - Fernandez IJ, Piccin O, Sciascia S, Cavicchi O, Repaci A, Vicennati V, Fiorentino M. Clinical significance of BRAF mutation in thyroid papillary cancer. Otolaryngol Head Neck Surg. 2013;148(6):919-925.
doi pubmed - Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, Nolop K, et al. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid. 2013;23(10):1277-1283.
doi pubmed - Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Dual inhibition of mitogen-activated protein kinase kinase and mammalian target of rapamycin in differentiated and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009;94(10):4107-4112.
doi pubmed - Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, Gilbert J, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18(7):2056-2065.
doi pubmed - Gupta-Abramson V, Troxel AB, Nellore A, Puttaswamy K, Redlinger M, Ransone K, Mandel SJ, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol. 2008;26(29):4714-4719.
doi pubmed - Kloos RT, Ringel MD, Knopp MV, Hall NC, King M, Stevens R, Liang J, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol. 2009;27(10):1675-1684.
doi pubmed - Ahmed M, Barbachano Y, Riddell A, Hickey J, Newbold KL, Viros A, Harrington KJ, et al. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol. 2011;165(2):315-322.
doi pubmed - Hong DS, Cabanillas ME, Wheler J, Naing A, Tsimberidou AM, Ye L, Busaidy NL, et al. Inhibition of the Ras/Raf/MEK/ERK and RET kinase pathways with the combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in medullary and differentiated thyroid malignancies. J Clin Endocrinol Metab. 2011;96(4):997-1005.
doi pubmed - Cabanillas ME, Waguespack SG, Bronstein Y, Williams MD, Feng L, Hernandez M, Lopez A, et al. Treatment with tyrosine kinase inhibitors for patients with differentiated thyroid cancer: the M. D. Anderson experience. J Clin Endocrinol Metab. 2010;95(6):2588-2595.
doi pubmed - Shen Y, Ruan M, Luo Q, Yu Y, Lu H, Zhu R, Chen L. Brain metastasis from follicular thyroid carcinoma: treatment with sorafenib. Thyroid. 2012;22(8):856-860.
doi pubmed - Ciappuccini R, Trzepla G, Heutte N, Sevin E, Galais MP, Bardet S. Sorafenib increases 18-FDG colic uptake: demonstration in patients with differentiated thyroid cancer. EJNMMI Res. 2012;2(1):18.
doi pubmed - Bible KC, Suman VJ, Molina JR, Smallridge RC, Maples WJ, Menefee ME, Rubin J, et al. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11(10):962-972.
doi - Bible KC, Suman VJ, Menefee ME, Smallridge RC, Molina JR, Maples WJ, Karlin NJ, et al. A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. J Clin Endocrinol Metab. 2012;97(9):3179-3184.
doi pubmed - Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, Kane MA, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26(29):4708-4713.
doi pubmed - Carr LL, Mankoff DA, Goulart BH, Eaton KD, Capell PT, Kell EM, Bauman JE, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16(21):5260-5268.
doi pubmed - Cleary JM, Sadow PM, Randolph GW, Palmer EL, Lynch TP, Nikiforov YE, Wirth LJ. Neoadjuvant treatment of unresectable medullary thyroid cancer with sunitinib. J Clin Oncol. 2010;28(23):e390-392.
doi pubmed - Gori S, Foglietta J, Rossi M, Hamzaj A, Stocchi L, Galuppo C, Picece V, et al. Sunitinib therapy in metastatic papillary thyroid cancer. Tumori. 2013;99(6):285e-287e.
pubmed - Ravaud A, de la Fouchardiere C, Asselineau J, Delord JP, Do Cao C, Niccoli P, Rodien P, et al. Efficacy of sunitinib in advanced medullary thyroid carcinoma: intermediate results of phase II THYSU. Oncologist. 2010;15(2):212-213; author reply 214.
doi pubmed - Sweeney CJ, Chiorean EG, Verschraegen CF, Lee FC, Jones S, Royce M, Tye L, et al. A phase I study of sunitinib plus capecitabine in patients with advanced solid tumors. J Clin Oncol. 2010;28(29):4513-4520.
doi pubmed - Sherman SI, Wirth LJ, Droz JP, Hofmann M, Bastholt L, Martins RG, Licitra L, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med. 2008;359(1):31-42.
doi pubmed - Schlumberger MJ, Elisei R, Bastholt L, Wirth LJ, Martins RG, Locati LD, Jarzab B, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol. 2009;27(23):3794-3801.
doi pubmed - Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, de la Fouchardiere C, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319-328.
doi - Coulter DW, Walko C, Patel J, Moats-Staats BM, McFadden A, Smith SV, Khan WA, et al. Valproic acid reduces the tolerability of temsirolimus in children and adolescents with solid tumors. Anticancer Drugs. 2013;24(4):415-421.
doi pubmed - Lim SM, Chang H, Yoon MJ, Hong YK, Kim H, Chung WY, Park CS, et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann Oncol. 2013;24(12):3089-3094.
doi pubmed - Druce M, Chung TT, Grozinsky-Glasberg S, Gross DJ, Grossman AB. Preliminary report of the use of everolimus in a patient with progressive medullary thyroid carcinoma. Clin Endocrinol (Oxf). 2012;77(1):154-155.
doi pubmed - Wunderlich A, Arndt T, Fischer M, Roth S, Ramaswamy A, Greene BH, Brendel C, et al. Targeting the proteasome as a promising therapeutic strategy in thyroid cancer. J Surg Oncol. 2012;105(4):357-364.
doi pubmed - Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139-2146.
doi pubmed - Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, Pentlow KS, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623-632.
doi pubmed - Hoftijzer H, Heemstra KA, Morreau H, Stokkel MP, Corssmit EP, Gelderblom H, Weijers K, et al. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol. 2009;161(6):923-931.
doi pubmed - Schutz FA, Choueiri TK, Sternberg CN. Pazopanib: Clinical development of a potent anti-angiogenic drug. Crit Rev Oncol Hematol. 2011;77(3):163-171.
doi pubmed - Bible KC, Suman VJ, Molina JR, Smallridge RC, Maples WJ, Menefee ME, Rubin J, et al. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab. 2014;99(5):1687-1693.
doi pubmed - Fujiwara Y, Kiyota N, Chayahara N, Suzuki A, Umeyama Y, Mukohara T, Minami H. Management of axitinib (AG-013736)-induced fatigue and thyroid dysfunction, and predictive biomarkers of axitinib exposure: results from phase I studies in Japanese patients. Invest New Drugs. 2012;30(3):1055-1064.
doi pubmed - Kelly RJ, Rixe O. Axitinib (AG-013736). Recent Results Cancer Res. 2010;184:33-44.
doi pubmed - Bass MB, Sherman SI, Schlumberger MJ, Davis MT, Kivman L, Khoo HM, Notari KH, et al. Biomarkers as predictors of response to treatment with motesanib in patients with progressive advanced thyroid cancer. J Clin Endocrinol Metab. 2010;95(11):5018-5027.
doi pubmed - Fury MG, Sherman E, Haque S, Korte S, Lisa D, Shen R, Wu N, et al. A phase I study of daily everolimus plus low-dose weekly cisplatin for patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;69(3):591-598.
doi pubmed - Putzer D, Gabriel M, Kroiss A, Madleitner R, Eisterer W, Kendler D, Uprimny C, et al. First experience with proteasome inhibitor treatment of radioiodine nonavid thyroid cancer using bortezomib. Clin Nucl Med. 2012;37(6):539-544.
doi pubmed - Harvey RD, Owonikoko TK, Lewis CM, Akintayo A, Chen Z, Tighiouart M, Ramalingam SS, et al. A phase 1 Bayesian dose selection study of bortezomib and sunitinib in patients with refractory solid tumor malignancies. Br J Cancer. 2013;108(4):762-765.
doi pubmed - Pennell NA, Daniels GH, Haddad RI, Ross DS, Evans T, Wirth LJ, Fidias PH, et al. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid. 2008;18(3):317-323.
doi pubmed - Anderson RT, Linnehan JE, Tongbram V, Keating K, Wirth LJ. Clinical, safety, and economic evidence in radioactive iodine-refractory differentiated thyroid cancer: a systematic literature review. Thyroid. 2013;23(4):392-407.
doi pubmed - Wells SA, Jr., Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134-141.
doi pubmed - Robinson BG, Paz-Ares L, Krebs A, Vasselli J, Haddad R. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2010;95(6):2664-2671.
doi pubmed - Leboulleux S, Bastholt L, Krause T, de la Fouchardiere C, Tennvall J, Awada A, Gomez JM, et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2012;13(9):897-905.
doi - Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, Pfister DG, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660-2666.
doi pubmed - Traynor K. Cabozantinib approved for advanced medullary thyroid cancer. Am J Health Syst Pharm. 2013;70(2):88.
doi - Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, Licitra L, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639-3646.
doi pubmed - Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, Cohen B, et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer. 2013;109(4):943-949.
doi pubmed - Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773-781.
doi - Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, Legos JJ, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res. 2012;18(8):2326-2335.
doi pubmed - Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893-1901.
doi - Stjepanovic N, Capdevila J. Multikinase inhibitors in the treatment of thyroid cancer: specific role of lenvatinib. Biologics. 2014;8:129-139.
pubmed - Ha HT, Lee JS, Urba S, Koenig RJ, Sisson J, Giordano T, Worden FP. A phase II study of imatinib in patients with advanced anaplastic thyroid cancer. Thyroid. 2010;20(9):975-980.
doi pubmed - de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ, de Vries MM, Links TP, Lips CJ, Voest EE. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92(9):3466-3469.
doi pubmed - Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, Rafie N, et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002;62(12):3408-3416.
pubmed - Pratilas CA, Xing F, Solit DB. Targeting oncogenic BRAF in human cancer. Curr Top Microbiol Immunol. 2012;355:83-98.
doi pubmed - Mooney CJ, Nagaiah G, Fu P, Wasman JK, Cooney MM, Savvides PS, Bokar JA, et al. A phase II trial of fosbretabulin in advanced anaplastic thyroid carcinoma and correlation of baseline serum-soluble intracellular adhesion molecule-1 with outcome. Thyroid. 2009;19(3):233-240.
doi pubmed - Hsieh AC, Truitt ML, Ruggero D. Oncogenic AKTivation of translation as a therapeutic target. Br J Cancer. 2011;105(3):329-336.
doi pubmed - Abbosh PH, Nephew KP. Multiple signaling pathways converge on beta-catenin in thyroid cancer. Thyroid. 2005;15(6):551-561.
doi pubmed - Burrows N, Babur M, Resch J, Ridsdale S, Mejin M, Rowling EJ, Brabant G, et al. GDC-0941 inhibits metastatic characteristics of thyroid carcinomas by targeting both the phosphoinositide-3 kinase (PI3K) and hypoxia-inducible factor-1alpha (HIF-1alpha) pathways. J Clin Endocrinol Metab. 2011;96(12):E1934-1943.
doi pubmed - Guigon CJ, Zhao L, Willingham MC, Cheng SY. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28(4):509-517.
doi pubmed - Sale EM, Hodgkinson CP, Jones NP, Sale GJ. A new strategy for studying protein kinase B and its three isoforms. Role of protein kinase B in phosphorylating glycogen synthase kinase-3, tuberin, WNK1, and ATP citrate lyase. Biochemistry. 2006;45(1):213-223.
doi pubmed - Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3(4):366-375.
doi pubmed - Xin M, Deng X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J Biol Chem. 2005;280(11):10781-10789.
doi pubmed - Maekawa T, Maniwa Y, Doi T, Nishio W, Yoshimura M, Ohbayashi C, Hayashi Y, et al. Expression and localization of FOXO1 in non-small cell lung cancer. Oncol Rep. 2009;22(1):57-64.
pubmed - Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28(12):4104-4115.
doi pubmed - Murugan AK, Alzahrani A, Xing M. Mutations in critical domains confer the human mTOR gene strong tumorigenicity. J Biol Chem. 2013;288(9):6511-6521.
doi pubmed - Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, Park IC, et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006;66(17):8511-8519.
doi pubmed - Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, Vasko V, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab. 2008;93(8):3106-3116.
doi pubmed - Abubaker J, Jehan Z, Bavi P, Sultana M, Al-Harbi S, Ibrahim M, Al-Nuaim A, et al. Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J Clin Endocrinol Metab. 2008;93(2):611-618.
doi pubmed - Liu B, Kuang A. [Genetic alterations in MAPK and PI3K/Akt signaling pathways and the generation, progression, diagnosis and therapy of thyroid cancer]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2012;29(6):1221-1225.
pubmed - Xing M. Identifying genetic alterations in poorly differentiated thyroid cancer: a rewarding pursuit. J Clin Endocrinol Metab. 2009;94(12):4661-4664.
doi pubmed - Kandil E, Tsumagari K, Ma J, Abd Elmageed ZY, Li X, Slakey D, Mondal D, et al. Synergistic inhibition of thyroid cancer by suppressing MAPK/PI3K/AKT pathways. J Surg Res. 2013;184(2):898-906.
doi pubmed - Burrows N, Telfer B, Brabant G, Williams KJ. Inhibiting the phosphatidylinositide 3-kinase pathway blocks radiation-induced metastasis associated with Rho-GTPase and Hypoxia-inducible factor-1 activity. Radiother Oncol. 2013;108(3):548-553.
doi pubmed - Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):121-130.
doi pubmed - Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. 2012;246(1):239-253.
doi pubmed - Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246(1):125-140.
doi pubmed - Pacifico F, Leonardi A. Role of NF-kappaB in thyroid cancer. Mol Cell Endocrinol. 2010;321(1):29-35.
doi pubmed - DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379-400.
doi pubmed - Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood. 2012;120(26):5188-5198.
doi pubmed - Storz P. Targeting the alternative NF-kappaB pathway in pancreatic cancer: a new direction for therapy? Expert Rev Anticancer Ther. 2013;13(5):501-504.
doi pubmed - Li L, Zhao F, Lu J, Li T, Yang H, Wu C, Liu Y. Notch-1 signaling promotes the malignant features of human breast cancer through NF-kappaB activation. PLoS One. 2014;9(4):e95912.
doi pubmed - Li X, Abdel-Mageed AB, Mondal D, Kandil E. The nuclear factor kappa-B signaling pathway as a therapeutic target against thyroid cancers. Thyroid. 2013;23(2):209-218.
doi pubmed - Liu J, Brown RE. Morphoproteomic confirmation of an activated nuclear factor-small ka, CyrillicBp65 pathway in follicular thyroid carcinoma. Int J Clin Exp Pathol. 2012;5(3):216-223.
pubmed - Pyo JS, Kang G, Kim DH, Chae SW, Park C, Kim K, Do SI, et al. Activation of nuclear factor-kappaB contributes to growth and aggressiveness of papillary thyroid carcinoma. Pathol Res Pract. 2013;209(4):228-232.
doi pubmed - Zhu W, Ou Y, Li Y, Xiao R, Shu M, Zhou Y, Xie J, et al. A small-molecule triptolide suppresses angiogenesis and invasion of human anaplastic thyroid carcinoma cells via down-regulation of the nuclear factor-kappa B pathway. Mol Pharmacol. 2009;75(4):812-819.
doi pubmed - Palona I, Namba H, Mitsutake N, Starenki D, Podtcheko A, Sedliarou I, Ohtsuru A, et al. BRAFV600E promotes invasiveness of thyroid cancer cells through nuclear factor kappaB activation. Endocrinology. 2006;147(12):5699-5707.
doi pubmed - Bommarito A, Richiusa P, Carissimi E, Pizzolanti G, Rodolico V, Zito G, Criscimanna A, et al. BRAFV600E mutation, TIMP-1 upregulation, and NF-kappaB activation: closing the loop on the papillary thyroid cancer trilogy. Endocr Relat Cancer. 2011;18(6):669-685.
doi pubmed - Yamashita AS, Geraldo MV, Fuziwara CS, Kulcsar MA, Friguglietti CU, da Costa RB, Baia GS, et al. Notch pathway is activated by MAPK signaling and influences papillary thyroid cancer proliferation. Transl Oncol. 2013;6(2):197-205.
doi pubmed - Komorowski J, Pasieka Z, Jankiewicz-Wika J, Stepien H. Matrix metalloproteinases, tissue inhibitors of matrix metalloproteinases and angiogenic cytokines in peripheral blood of patients with thyroid cancer. Thyroid. 2002;12(8):655-662.
doi pubmed - Bauerle KT, Schweppe RE, Haugen BR. Inhibition of nuclear factor-kappa B differentially affects thyroid cancer cell growth, apoptosis, and invasion. Mol Cancer. 2010;9:117.
doi pubmed - Altmann A, Markert A, Askoxylakis V, Schoning T, Jesenofsky R, Eisenhut M, Haberkorn U. Antitumor effects of proteasome inhibition in anaplastic thyroid carcinoma. J Nucl Med. 2012;53(11):1764-1771.
doi pubmed - Riesco-Eizaguirre G, Santisteban P. A perspective view of sodium iodide symporter research and its clinical implications. Eur J Endocrinol. 2006;155(4):495-512.
doi pubmed - Garcia-Jimenez C, Santisteban P. TSH signalling and cancer. Arq Bras Endocrinol Metabol. 2007;51(5):654-671.
doi pubmed - Liu X, Gao M. [Correlation between thyroid-stimulating hormone-peroxiredoxin1 signaling pathway and invasion of papillary thyroid carcinoma]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2009;44(4):287-291.
pubmed - Wang ZF, Liu QJ, Liao SQ, Yang R, Ge T, He X, Tian CP, et al. Expression and correlation of sodium/iodide symporter and thyroid stimulating hormone receptor in human thyroid carcinoma. Tumori. 2011;97(4):540-546.
pubmed - D'Agostino M, Sponziello M, Puppin C, Celano M, Maggisano V, Baldan F, Biffoni M, et al. Different expression of TSH receptor and NIS genes in thyroid cancer: role of epigenetics. J Mol Endocrinol. 2014;52(2):121-131.
doi pubmed - Matsumoto H, Sakamoto A, Fujiwara M, Yano Y, Shishido-Hara Y, Fujioka Y, Kamma H. Decreased expression of the thyroid-stimulating hormone receptor in poorly-differentiated carcinoma of the thyroid. Oncol Rep. 2008;19(6):1405-1411.
pubmed - Xing M, Usadel H, Cohen Y, Tokumaru Y, Guo Z, Westra WB, Tong BC, et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res. 2003;63(9):2316-2321.
pubmed - Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107-116.
doi pubmed - Calebiro D, de Filippis T, Lucchi S, Covino C, Panigone S, Beck-Peccoz P, Dunlap D, et al. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum Mol Genet. 2005;14(20):2991-3002.
doi pubmed - Fricke-Otto S, Pfarr N, Muhlenberg R, Pohlenz J. Mild congenital primary hypothyroidism in a Turkish family caused by a homozygous missense thyrotropin receptor (TSHR) gene mutation (A593 V). Exp Clin Endocrinol Diabetes. 2005;113(10):582-585.
doi pubmed - Coelho SM, Vaisman M, Carvalho DP. Tumour re-differentiation effect of retinoic acid: a novel therapeutic approach for advanced thyroid cancer. Curr Pharm Des. 2005;11(19):2525-2531.
doi pubmed - Jeong H, Kim YR, Kim KN, Choe JG, Chung JK, Kim MK. Effect of all-trans retinoic acid on sodium/iodide symporter expression, radioiodine uptake and gene expression profiles in a human anaplastic thyroid carcinoma cell line. Nucl Med Biol. 2006;33(7):875-882.
doi pubmed - Zarnegar R, Brunaud L, Kanauchi H, Wong M, Fung M, Ginzinger D, Duh QY, et al. Increasing the effectiveness of radioactive iodine therapy in the treatment of thyroid cancer using Trichostatin A, a histone deacetylase inhibitor. Surgery. 2002;132(6):984-990; discussion 990.
doi pubmed - Hou P, Bojdani E, Xing M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J Clin Endocrinol Metab. 2010;95(2):820-828.
doi pubmed - Fenton MS, Marion KM, Salem AK, Hogen R, Naeim F, Hershman JM. Sunitinib inhibits MEK/ERK and SAPK/JNK pathways and increases sodium/iodide symporter expression in papillary thyroid cancer. Thyroid. 2010;20(9):965-974.
doi pubmed - Kogai T, Brent GA. The sodium iodide symporter (NIS): regulation and approaches to targeting for cancer therapeutics. Pharmacol Ther. 2012;135(3):355-370.
doi pubmed - Xing Y, Clements WK, Kimelman D, Xu W. Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex. Genes Dev. 2003;17(22):2753-2764.
doi pubmed - Xu HT, Wei Q, Liu Y, Yang LH, Dai SD, Han Y, Yu JH, et al. Overexpression of axin downregulates TCF-4 and inhibits the development of lung cancer. Ann Surg Oncol. 2007;14(11):3251-3259.
doi pubmed - Kurihara T, Ikeda S, Ishizaki Y, Fujimori M, Tokumoto N, Hirata Y, Ozaki S, et al. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid. 2004;14(12):1020-1029.
doi pubmed - Zhang J, Gill AJ, Issacs JD, Atmore B, Johns A, Delbridge LW, Lai R, et al. The Wnt/beta-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum Pathol. 2012;43(7):1044-1050.
doi pubmed - DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69(13):5364-5373.
doi pubmed - Cho SW, Lee EJ, Kim H, Kim SH, Ahn HY, Kim YA, Yi KH, et al. Dickkopf-1 inhibits thyroid cancer cell survival and migration through regulation of beta-catenin/E-cadherin signaling. Mol Cell Endocrinol. 2013;366(1):90-98.
doi pubmed - Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W, Park JK, et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr Cancer Drug Targets. 2004;4(8):653-671.
doi pubmed - Rao AS, Kremenevskaja N, von Wasielewski R, Jakubcakova V, Kant S, Resch J, Brabant G. Wnt/beta-catenin signaling mediates antineoplastic effects of imatinib mesylate (gleevec) in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2006;91(1):159-168.
doi pubmed - Greenwald I, Kovall R. Notch signaling: genetics and structure. WormBook. 2013:1-28.
doi pubmed - Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13(9):654-666.
doi pubmed - Hu YY, Zheng MH, Zhang R, Liang YM, Han H. Notch signaling pathway and cancer metastasis. Adv Exp Med Biol. 2012;727:186-198.
doi pubmed - Takebe N, Nguyen D, Yang SX. Targeting notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther. 2014;141(2):140-149.
doi pubmed - Ferretti E, Tosi E, Po A, Scipioni A, Morisi R, Espinola MS, Russo D, et al. Notch signaling is involved in expression of thyrocyte differentiation markers and is down-regulated in thyroid tumors. J Clin Endocrinol Metab. 2008;93(10):4080-4087.
doi pubmed - Yu XM, Phan T, Patel PN, Jaskula-Sztul R, Chen H. Chrysin activates Notch1 signaling and suppresses tumor growth of anaplastic thyroid carcinoma in vitro and in vivo. Cancer. 2013;119(4):774-781.
doi pubmed - Jaskula-Sztul R, Pisarnturakit P, Landowski M, Chen H, Kunnimalaiyaan M. Expression of the active Notch1 decreases MTC tumor growth in vivo. J Surg Res. 2011;171(1):23-27.
doi pubmed - Greenblatt DY, Cayo MA, Adler JT, Ning L, Haymart MR, Kunnimalaiyaan M, Chen H. Valproic acid activates Notch1 signaling and induces apoptosis in medullary thyroid cancer cells. Ann Surg. 2008;247(6):1036-1040.
doi pubmed - Patel PN, Yu XM, Jaskula-Sztul R, Chen H. Hesperetin activates the Notch1 signaling cascade, causes apoptosis, and induces cellular differentiation in anaplastic thyroid cancer. Ann Surg Oncol. 2014;21(Suppl 4):S497-504.
doi pubmed - Lee SJ, Lee MH, Kim DW, Lee S, Huang S, Ryu MJ, Kim YK, et al. Cross-regulation between oncogenic BRAF(V600E) kinase and the MST1 pathway in papillary thyroid carcinoma. PLoS One. 2011;6(1):e16180.
doi pubmed - Chattopadhyay C, El-Naggar AK, Williams MD, Clayman GL. Small molecule c-MET inhibitor PHA665752: effect on cell growth and motility in papillary thyroid carcinoma. Head Neck. 2008;30(8):991-1000.
doi pubmed - Liu X, Newton RC, Scherle PA. Development of c-MET pathway inhibitors. Expert Opin Investig Drugs. 2011;20(9):1225-1241.
doi pubmed - Xu X, Ding H, Rao G, Arora S, Saclarides CP, Esparaz J, Gattuso P, et al. Activation of the Sonic Hedgehog pathway in thyroid neoplasms and its potential role in tumor cell proliferation. Endocr Relat Cancer. 2012;19(2):167-179.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.