

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 7, Number 10, October 2015, pages 742-751
Aurora Kinases and Potential Medical Applications of Aurora Kinase Inhibitors: A Review
Paschalis Gavriilidisa, c, Alexandros Giakoustidisb, Dimitrios Giakoustidisb
aDepartment of Surgical Oncology, Theageneio Anticancer Hospital, Thessaloniki, Greece
bDepartment of Transplantation Surgery, Hippokrateion University Hospital, Thessaloniki, Greece
cCorresponding Author: Paschalis Gavriilidis, Department of Surgical Oncology, Theageneio Anticancer Hospital, Thessaloniki, Greece
Manuscript accepted for publication August 11, 2015
Short title: Aurora Kinases and AKIs
doi: http://dx.doi.org/10.14740/jocmr2295w
- Abstract
- Introduction
- Aurora A
- Aurora B
- Aurora C
- Potential Oncological Applications
- Pan-Aurora Inhibitors
- Selective AKIs
- Combined Use of AKIs With Histone Deacetylase (HDAC) Inhibitors
- The Role of AKs in GIST and Head and Neck Cancers
- Conclusions
- References
| Abstract | ▴Top |
Aurora kinases (AKs) represent a novel group of serine/threonine kinases. They were originally described in 1995 by David Glover in the course of studies of mutant alleles characterized with unusual spindle pole configuration in Drosophila melanogaster. Thus far, three AKs A, B, and C have been discovered in human healthy and neoplastic cells. Each one locates in different subcellular locations and they are all nuclear proteins. AKs are playing an essential role in mitotic events such as monitoring of the mitotic checkpoint, creation of bipolar mitotic spindle and alignment of centrosomes on it, also regulating centrosome separation, bio-orientation of chromosomes and cytokinesis. Any inactivation of them can have catastrophic consequences on mitotic events of spindle formation, alignment of centrosomes and cytokinesis, resulting in apoptosis. Overexpression of AKs has been detected in diverse solid and hematological cancers and has been linked with a dismal prognosis. After discovery and identification of the first aurora kinase inhibitor (AKI) ZM447439 as a potential drug for targeted therapy in cancer treatment, approximately 30 AKIs have been introduced in cancer treatment.
Keywords: Serine/threonine kinases; Aurora kinases A, B and C; Aurora kinase inhibitors
| Introduction | ▴Top |
Living organisms grow, regenerate and inherit their genetic material with the help of the cell cycle process. As a result of the ordered events of the cell cycle, two daughter cells appear with two exact halves of genetic material. The principal characteristic of cell division is precise division of genetic material. The replication of chromosomal DNA takes place in the S phase of the cell cycle. Segregation of chromosomes and maintenance of diploid chromosome content is completed during mitosis; vast numbers of proteins precisely tune and smoothly regulate this process. Mitotic kinases are essential regulators of mechanisms of protein localization, proteolysis and phosphorylation and contribute for a smooth progression through mitosis [1, 2]. During mitosis, microtubules, the building material of the mitotic spindle and the centrosome, play an important role in regulating tubulin dynamics [3]. Any errors in mitotic signaling pathways can lead to uncontrolled proliferation, one of the principal characteristics of tumors.
Intervention and regulation of mitotic events are the pillars of contemporary chemotherapy. The main flaw of the current antimitotic drugs is that they target simultaneously proliferating cancer cells and non-proliferating healthy cells leading to side effects [3, 4]. Therefore, researchers are trying to identify more specific mitotic targets. Lately, cyclin-dependent kinases, survivin, polo-kinase and aurora kinases (AKs) have been proposed as targets for anticancer therapy [5-7].
The first human homologue of AKs was described by Bischoff and Zhou in 1998 [4, 8]. So far in human cells, three AKs were identified: A, B and C [9]. AKs structure has been conserved during eukaryotic evolution [1]. They comprise two domains: the NH2-terminal regulatory domain and the COOH-terminal catalytic domain. All three share great homology in the catalytic domain but differ in the regulatory [10, 11] (Fig. 1). In 2004, the first patient was treated with aurora kinase inhibitor (AKI) (PHA-739358) [12]. The translational period was only 8 years. Comparing with the translational period of other targeted therapies, we can remark a real progress.
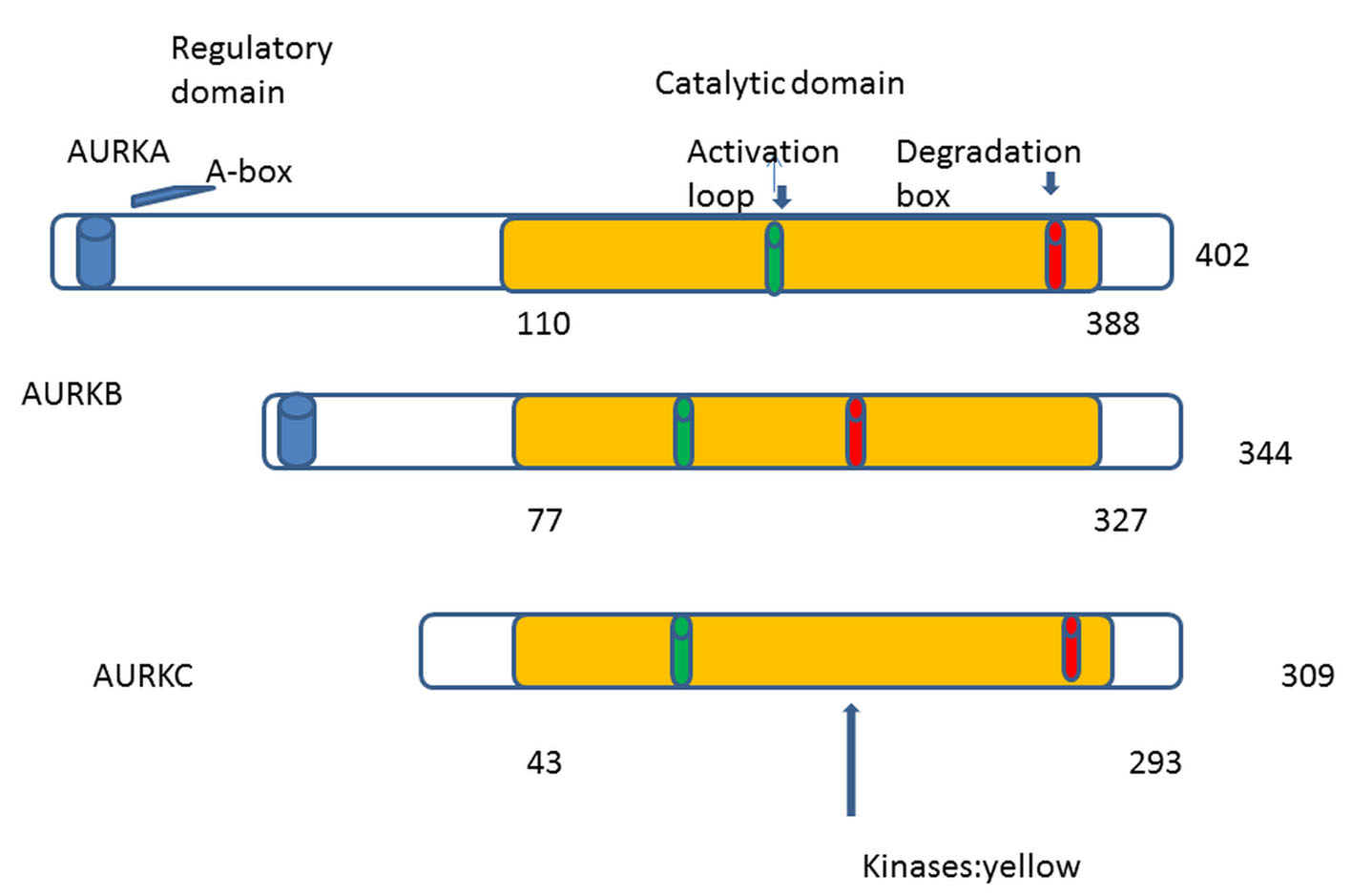 Click for large image | Figure 1. AKs contain mainly two domains: 1) NH2-terminal regulatory domain (white), 2) COOH-terminal catalytic domain (yellow). The three auroras A, B, and C share great homology in the catalytic domain. Phosphorylation at threonine within the activation loop is necessary for kinase activity. |
| Aurora A | ▴Top |
Each AK maps in a different chromosome, occupies particular subcellular localizations, turns over and regulates different cell cycle events.
Aurora A maps to chromosome 20q13.2; its activity is synchronized by phosphorylation and dephosphorylation as well as by its expression and degradation. Aurora A is degraded or inactivated in the G1 phase of the cell cycle and in contrast to G2/M phases, it expresses its maximal activity. It is persistently expressed and strictly regulates mitotic events from mitotic entry through mitotic exit including centrosome maturation, centrosome separation, bipolar spindle assembly, chromosome alignment and cytokinesis [13-15].
Aurora A is considered a bona fide proto-oncogene and the interest of researchers was intensified when first aurora A was found to be overexpressed in human breast cancer cell lines and so named breast tumor activated kinase (BTAK) and in primary colorectal carcinoma biopsies. Consequently, they observed the ability of overexpressed aurora A to transform rodent cells NIH3T3 and rat1 and to provoke tumorigenesis in nude mice [16-19].
Goepfert et al observed in early stages of oncogenesis of the rat breast cancer model an overexpression of aurora A and centrosome amplification [17]. In 2007, Nishida et al showed that overexpression of the aurora A gene is related with chromosomal instability in colorectal cancers [18]. It has also been reported that overexpression and amplification of aurora A in hepatocellular and gastric cancer is a marker of aneuploidy and dismal prognosis [19, 20].
Normal growth rate of cells, cell viability and oncogenesis depend on interactions and balances between aurora A and tumor suppressor gene products such as Chfr, BRCA1, and p53 [21-24]. Chfr is a known tumor suppressor and a mitotic checkpoint protein that confirms chromosomal stability by interacting and influencing expression of aurora A [21]. Ouchi et al showed that aurora A connects to BRCA1 and phosphorylates Ser 308. They also suggested that disrupted regulation of G2/M transition by BRCA1 and aurora A predisposes to carcinogenesis [22]. Aurora A is an important regulatory constituent of the p53 pathway and interacts with it at multiple levels. It is overexpression inhibits the p53 suppression function, which facilitates oncogenesis [23]. Moreover, aurora A interacts with p53 and influences its suppression function by at least two ways: firstly it directly phosphorylates p53 at Ser 315 enabling mdm2 mediated degradation of p53 in tumor cells and secondly it inactivates its transcriptional activity by phosphorylating p53 at Ser 215 [23, 24]. It is suggested that aurora A, as a constituent to the Ras/Raf/MEK/ERK/Map kinase pathway of a human carcinogenesis, might influence the growth rate of cancer cells, and also stimulates telomerase activity and human telomerase reverse transcriptase by up-regulation of c-myc23 and promotes collagen I-induced cell migration [25]. Therefore aurora A might promise prolonged survival in cancer cells.
Overexpression of aurora A plays a crucial role in the inactivation of apoptosis in cancer cells by the up-regulation of an anti-apoptotic effector NF-kB [26].
| Aurora B | ▴Top |
Aurora B maps to chromosome 17q13. Independently and of great similarity in structure and sequence, aurora A and B are totally different in subcellular localizations; their subcellular geography determines independent functions in mitotic events. During mitosis and when prometaphase starts, aurora B leaves the nucleus and relocates to centromeres until metaphase; once anaphase begins, it moves steadily to the midzone and remains at the midbody until the end of cytokinesis [27].
During mitosis, aurora B forms a tight complex with its binding partners and substrates such as: inner centrosome protein (INCEP), borealin and survivin [26]. This complex called chromosomal passenger complex plays an essential role in the establishment of proper kinetochore-microtubule attachments [28, 29]. Aurora B also assists in proper chromosome bio-orientation, accurate chromosomal segregation, regulation of the mitotic checkpoint and cytokinesis [30].
While inhibition of aurora A leads to arrest in mitosis, in contrast inhibition of aurora B inactivates the checkpoints and leads to chromosome misalignment and failed cytokinesis [31]. Owing to this characteristic, aurora B inhibitors have been named as mitotic drivers in a review [32].
Multinucleation and polyploidy are consequences of aurora B overexpression; in addition, these phenotypes are aggravated in absence of p53 [33]. Moreover, overexpressed aurora B causes chromosome insulation in metaphase, defects in chromosome segregation and defects in cytokinesis, so inducing the process of carcinogenesis [34].
Aurora B is overexpressed in hepatocellular, colorectal, breast, renal, lung and thyroid cancer [35-37]. It is reported that overexpressed aurora B is an essential marker related with tumor invasiveness, early recurrence and dismal prognosis [38].
Inappropriate cell movement that underlies tumor invasive and metastatic possibility are promoted by both AKs A and B [39].
Yasen et al [40] studying the role and expression of aurora B and its variants, wild type, splicing variant 1 and 2 in hepatocarcinogenesis and in liver tumors concluded that aurora B splicing variant 2 (SV2) was associated with hepatocellular cancers (HCCs) diagnosed with vascular invasion of hepatic and portal veins. Therefore, there is justified evidence to correlate this variant with dismal prognosis and high rate of recurrence in HCC. They also detected SV2 to be overexpressed in cases of multiple tumor formation, capsular invasion and increased AFP levels. Characteristically, SV2 was not detected in normal hepatic tissues but only in neoplastic tissues. Based on these features, inhibition of aurora B and its variants might provide useful tools for neoadjuvant and adjuvant treatment of the HCC in the near future.
| Aurora C | ▴Top |
Aurora C is the less investigated aurora compared to AKs A and B. It maps to chromosome 19q13. It was first discovered in testis where it functions in spermatogenesis [41].
Structurally, it is a chromosomal passenger protein like aurora B and is more similar to it (83%) than to aurora A (71%). It plays an essential role in mitotic events and in particular in centrosome function and is identified to it from anaphase to cytokinesis [41]. Similar to aurora B, its mRNA and protein expression levels peak at G2/M phases [41].
Studying the role of aurora C on female mouse meiosis, researchers detected that aurora C inactivation brings about defects such as premature chromosome segregation, chromosome misalignment, abnormal kinetochore-microtubule attachment and cytokinesis in meiosis [42].
It is reported to be overexpressed in HeLa, HepG2, MDA-MD-453 and Hu H7 cancer cells [43]. However, there is no enough evidence for its role in carcinogenesis and further research is required to clarify its role in molecular pathways in cancer.
Aurora B and C are closely related and most likely when developed, form a common ancestral aurora B/C of cold-blooded vertebrate [44, 45].
| Potential Oncological Applications | ▴Top |
Thus far, many studies found that overexpressed and amplified AKs are tightly associated with a large number of cancers. Due to their participation in mitotic events and influence on important processes of human carcinogenesis such as Ras/Raf/MEK/ERK/MAP kinase pathway, telomerase activity, collagen I-induced cell migration and anchorage-independent growth, they trigger interest for researchers to develop potential inhibitors against them (Table 1).
 Click to view | Table 1. Potential Oncological Applications |
| Pan-Aurora Inhibitors | ▴Top |
Tozasertib
Tozasertib is a pyrimidine derivative with high affinity to AKs, imatinib-resistant and dasatinib-resistant forms of ABL. Its inhibitory constant values are lower for aurora A 0.6 nM and C 4.6 nM than for aurora B 18 nM [46]. In-vitro showed a 100-fold more selectivity for AKs than for a pool of 55 other kinases. In-vivo demonstrated inhibitive possibilities of a large number of cancers. In particular, tumor xenograft models regressed and reduced the tumor size by 22-98% [47].
Tozasertib was the first AKI to be used in patients. Its main side effects were reversible neutropenia and anorexia. It was stopped in 2007 because QTc prolongation occurred in one patient. However, ASCO2009 reported its use in patients with chronic myelogenous leukemia (CML) and ALL in simultaneous and successive treatments with dasatinib [46].
Patients with a solid advanced tumor tolerated well tozasertib in doses up to 8 mg/m2/h in phase I study [48].
PHA-739358 (danusertib)
Danusertib is a potent pan-aurora kinase inhibitor; its half maximal inhibitory concentration (IC50) is obviously the smallest for aurora A only 13 nM, the highest for B 79 nM and for C 61 nM. Chemically it is a 3-aminopyrazole derivative [49]. It also inhibits several cancer related tyrosine kinases as well as Abl, Trk-a, fibroblast growth receptor-1 and Ret [49]. This feature of cross-reactivity enhances its activity and broadens its indications in hematological and solid malignancies such as CML, ALL, thyroid, prostate and breast cancer [49]. Danusertib has significant anti-tumor activity in transgenic models with a favorable preclinical safety profile; target organs of danusertib are hemolymphopoietic system, gastrointestinal tract, kidneys and male reproductive organs [49].
Benten et al observed that combination of multikinase inhibitor sorafenib and PHA-739358 caused a virtual growth arrest of hepatocellular cancers. In addition, standard monotherapy dose of PHA-739358 alone was capable of shrinking tumors even after treatment failure under sorafenib [50].
Fraedrich et al showed that danusertib inhibits advanced gastroenteropancreatic neuroendocrine tumor cell growth in association with cell-cycle disruption and induction of apoptosis [51].
Cohen et al [52] in a phase I study treated 56 patients with advanced solid tumors by dose escalating danusertib. Group I consisted of 40 patients and was treated by danusertib alone; the maximum tolerated dose (MTD) was 500 mg/m2. Group II consisted of 16 patients and was treated concomitantly with danusertib and granulocyte colony stimulating factor (G-CSF) filgrastim; the MDT was 750 mg/m2. In one patient of group II with diagnosis of refractory small cell lung cancer, objective tumor response was observed when he was treated with doses at 1,000 mg/m2. Subsequently the dose was reduced to 750 mg/m2 when azotemia occurred. They also observed disease stabilization in 43% of patients of group I and 50% of group II. The time period of 23.9 to 52.3 weeks was defined as being prolonged disease stabilization time and was only observed in four patients of group I. Tumor regression by 27% and declination CA-125 by 30% was achieved in a woman with ovarian cancer. The most frequently encountered serious side effects were leukopenia, neutropenia, febrile neutropenia, neutropenic infections, azotemia and pyrexia. They concluded that their results provide ample justification for further clinical trials [52].
Danusertib related renal side effects have been investigated in rats and dogs. In the rat model, tubular nephropathy was noticed at the 600 and 750 mg/m2 intravenous (IV) bolus equivalent doses. In repeated dose studies with 6-h IV infusions, an increase in urea and creatinine was observed in rats (but not in dogs) at the MTD 720 mg/m2 and was rescindable [52]. In man, an increase in grade 1 and 2 creatinine was seen when the administered dose was > 1,000 mg/m2 [52].
Steeghs et al enrolled 50 patients with advanced or metastatic solid tumors in a phase I study; in group I was given danusertib at starting dose 45 mg/m2 in 6-h IV infusion scheme, in group II 250 mg/m2 (IV) in 3-h scheme. On both groups danusertib was given on days 1, 8, and 15 every 4 weeks. They reported neutropenia as a main adverse effect on both groups. They did not observe partial or complete responses. Overall disease control was achieved only in six of 30 patients (20%) of group of 6-h infusion. Disease stabilized for more than 6 months in four patients in the 6-h scheme and in one in the 3-h scheme. Interestingly, disease stabilization was achieved in one patient with uncontrolled non-small cell lung cancer treated on the 6-h scheme before the study. Finally they concluded that the recommended dose of danusertib for phase II studies is 330 mg/m2 in 6-h IV infusion on days 1, 8, and 15 every 4 weeks [53].
Meulenbeld et al [54] studied the single agent efficacy of danusertib in patients with metastatic castration resistant prostate cancer after docetaxel failure. They concluded that danusertib monotherapy is ineffective.
Recently, danusertib proved to be effective on treating the neglected disease African sleeping sickness or human African trypanosomiasis. Ochiana et al demonstrated that danusertib inhibits trypanosomal aurora kinase 1 (TbAUK1) and kills the parasite in vitro [55].
CYC116
CYC116 is a pan-AKI which in preclinical studies demonstrated selective effects on aurora A and B. Its route of administration is oral and overall antitumor effect is based on inhibition of angiogenesis [56, 57].
It demonstrated characteristic activity against cancer models of both cell lines and murine xenografts of thyroid, breast, pancreatic, colorectal melanoma and non-small cell lung cancers [58, 59].
It is also demonstrated synergy when used in combination with chemotherapeutic agents and ionizing radiation [59]. The results of the preclinical study showed that the combined use of the CYC116 with ionizing radiation has stronger anti-tumor results on Ras-mutated colorectal adenocarcinoma than on Ras-wild type cell lines [59].
SNS-314
SNS is a pan-AKI expressing highest affinity for aurora B (IC50 = 31 nM) followed by aurora A (9 nM) and aurora C (3 nM). It is aminothiazole-derived urea; its anti-tumor effects for cancers of prostate, colon, breast, melanoma and ovary were demonstrated in preclinical studies of cell lines and murine models [60].
One of the principal characteristics of SNS314 is the exhibition of synergy when it is used in combined treatment schemes with other chemotherapeutic agents in colorectal cell lines; moreover with antimicrotubule agents, its synergistic effect is further enhanced [61].
AMG-900
It is a small molecule pan-aurora inhibitor orally administered and demonstrates characteristically little off-target inhibition [62]. Its anti-proliferative capacities are proved against cell lines of both hematologic and solid cancers. Furthermore, it proved to be effective in cell lines resistant to paclitaxel, AZD-1152, tozasertib and danusertib [62].
VE-465
VE-465 is an aurora inhibitor related to MK0457. Its antineoplastic activity was confirmed in preclinical studies of culture cells and murine xenograft models of hepatocellular carcinoma, ovarian cancer, multiple myeloma, myeloid leukemia and CML [63, 64].
AS703569/R-763
This pan-AKI inactivates aurora A, B and C at IC50 values of 4, 4.8 and 6.9 nM respectively. Furthermore, it inhibits oncogenic kinases including fms-related tyrosine kinase (FLT1), FLT3 and Abl [65].
It is an orally administered aurora inhibitor exhibiting effective wide inhibition of Akt, VEGFR3, FLT3, Bcr-Abl, and IGFR [65]. Its anti-proliferative activity was demonstrated in cell culture and murine xenograft model of hemopoietic and solid cancers including biphenotypic leukemia, acute promyelocytic leukemia, acute lymphocytic leukemia, CML, multiple myeloma, colorectal, ovarian, breast and pancreatic cancers [65, 66].
PF-03814735
PF-03814735 is another pan-aurora inhibitor expressing highest affinity for aurora A and B at IC50 of 6 and 0.8 nM, respectively. Moreover, at higher IC50 values, it inhibits fibroblast growth factor receptor 1 (FGFR1), focal adhesion kinase (FAK), TrKA, FLT1, and MET kinases [67]. A recent phase I study was conducted where 57 patients with solid tumors were enrolled. In arm A, 32 individuals were treated for 5 days with 5 - 100 mg/day; in arm B, 25 individuals were treated for 10 days with 40 - 60 mg/day of 21-day cycles. For the patients of arm A, 80 mg/day was the MTD. In this cohort of patients, two suffered grade 3 and grade 4 febrile neutropenia and one suffered from grade 3 proctalgia. In arm B, 50 mg was the MTD where two patients suffered from ventricular dysfunction and one grade 3 increase of aspartate aminotransferase. The substance was easily absorbed, appeared in 6 h and showed favorable linear pharmacokinetics.
Results wise, stabilization of the disease was observed in 19 patients. Furthermore, tumor reduction was observed in five patients of arm A with stable disease occurring [68].
AT9283
AT9283 has a wide range inhibitory effect at IC50 of 3 nM. It inhibits both aurora A and B and at IC50 of 110 and 3 nM, it inhibits T3151 BCR-ABL and wild-type BCR-ABL, respectively [69, 70]. Moreover it targets ABL, Janus kinase (JAK), JAK3 and restrains proliferation of BaF3 cells in both T3151 BCR-ABL and wild-type BCR-ABL [69, 70].
In K562 xenograft mouse model of BCR-ABL positive CML has been observed to reduce significantly tumor volume at dose 12.5 mg/kg [70].
In phase I trial where 30 patients with refractory leukemia were enrolled with a dose range of 3 - 162 mg/m2/day, dose limiting toxicity (DLT) was not observed up to dose 72 mg/m2/day. Only in one case at a dose of 12 mg/m2/day tumor lysis syndrome was observed. The most common developed DLTs were mucositis, septicemia and pneumonia [71].
GSK1070916
GSK1070916 selectively inhibits aurora B-INCEP and aurora C-INCEP. It interacts with tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE2), salt inducible kinase (SIK), FLT1, FGFR1 and FLT4 [72]. After testing its in-vitro activity on 161 tumor cell lines, inhibition of cancer lines at IC50 of 8 nM without showing any remarkable anticancer activity on non-proliferating HUVEC cells was observed [73].
In-vivo efficacy of the GSK1070916 has been examined in several xenograft models: in MCF-7, colo205 and H460. Stabilization of the process and partial or complete regression in K562, HCT116, HL60 and A549 models was observed [73].
| Selective AKIs | ▴Top |
MLN8054 (millennium)
This substance is a highly selective inhibitor of aurora A. It acts not by degradation or down-regulation of aurora A but inactivates its phosphorylation. It is > 40 folds more effective inhibitor of aurora A than of aurora B. However at higher concentrations it inhibits also aurora B. Abnormal DNA content in cells treated with millennium is observed [74].
Remarkable tumor growth inhibition occurred in tumor xenograft models treated with MLN8054. However the substance was withdrawn from clinical trials due to severe side effects including severe somnolence, central neurocognitive changes and central nervous system toxicity. This phenomenon was explained by the structural similarity of the MLN8054 to the benzodiazepines and associated affinity to gamma-aminobutyric acid a1 receptor (GABA a1) [75, 76].
MLN8237 (alisertib)
Millennium invented a second generation selective inhibitor of aurora A, the 5-H-pyrimidol[5,4-d] [2] benzazepine MLN8237 due to the demonstrated side effects of the MLN8054. In preclinical studies, inhibition of aurora A was achieved with an IC50 value of 1.2 nmol/L, while aurora B inhibition was achieved with a value of 396.5 nmol/L [9]. The demonstrated most common DLTs in phase I studies were thrombocytopenia, neutropenia, stomatitis and diarrhea [77, 78]. In-vivo antitumor activity of the drug in various xenograft tumor models was observed [79]. The substance has been found to be active against T3151 BCR-ABL and BCR-ABL mutated cells in culture [80]. MLN8237 has been examined on pediatric cancer cell lines such as glioblastoma, Ewing sarcoma, neuroblastoma, rhabdomyosarcoma, AML and ALL. The highest efficacy was demonstrated against the ALL cell lines and the least on rhabdomyosarcoma [81].
| Combined Use of AKIs With Histone Deacetylase (HDAC) Inhibitors | ▴Top |
Recently patients with advanced phase CML treated with BCR-ABL tyrosine kinase inhibitor imatinib, have developed resistance to treatment [82]. The cause of this phenomenon is based on point mutations in the kinase domain of BCR-ABL [83]. Researchers tried to find a solution to the problem by combining AKI tozasertib with HDAC inhibitors pracinostat and/or vorinostat. They concluded that the combination of AKI tozasertib and HDAC pracinostat and/or vorinostat is highly effective against BCR-ABL cells [84].
| The Role of AKs in GIST and Head and Neck Cancers | ▴Top |
AK overexpression was identified as a poor prognostic factor for GIST recurrence [85]. Moreover aurora A is significantly overexpressed in non-gastric GISTs than in gastric.
Lagarde et al demonstrated that AK A is strongly associated with GIST tumors with high metastatic potential [86]. Either aurora A measured by immunohistochemistry or by qRT-PCR both confirmed it as an independent prognostic factor for GIST recurrence [85, 86].
An essential prerequisite for oncogenesis in squamous-cell cancer head and neck cells (SCCHN) is the build-up of aurora A in the nucleus [87]. Using immunohistochemistry (IHC) and qRT-PCR, researchers measured aurora A mRNA and aurora A protein expression in 66 head and neck cancers and healthy squamous epithelium. They concluded that decreased disease free survival and overall survival was related to increased levels of aurora A mRNA [88]. Another retrospective study reported overexpression of aurora A mRNA in laryngeal cancer [89].
Qi et al demonstrated that aurora B overexpression in oral cancer is related with increased multinuclear cells, cellular proliferation and lymph node metastases [90].
Phase I trial patients with advanced solid tumors were treated with MLN8054. From the cohort of head and neck tumors, only a few of them demonstrated stabilization of the disease [75, 78].
New therapeutic strategies are investigating the combined use of the EGFR inhibitor cetuximab, AKIs and radiotherapy [91]. Simultaneous use of cetuximab and the AKI R763 has shown inhibition of cell growth in SCCHN cell lines [92]. It is also reported that overexpression of aurora A and EGFR are dismal prognostic factors [92].
| Conclusions | ▴Top |
Discovery of AKs provides a new concept to comprehend cell cycle events and especially mitosis which are affected on the beginning of carcinogenesis. Overexpression and/or amplification of AKs is found in a wide range of hematological and solid cancers. Aberrant auroras can lead to genomic instability and subsequently to uncontrolled cell proliferation which is the hallmark of cancer and the prerequisite for emerging accrual mutations which enable cancers to develop resistance to chemotherapeutic agents. Tumor cell sensitivity is highly dependent on intact cell cycle checkpoints. AK inhibition expresses a natural degree of selectivity to cancer cells depleted of a certain mitotic checkpoint proteins and non-functional p53 rather than to healthy tissues. The above feature promises more effective AKIs and fewer serious adverse effects.
Conflict of Interests
None.
Abbreviations
AK: aurora kinase; AKI: aurora kinase inhibitor; HDAC: histone deacetylase
| References | ▴Top |
- Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol. 2005;15(5):241-250.
doi pubmed - Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5(1):42-50.
doi pubmed - Schatten H. The mammalian centrosome and its functional significance. Histochem Cell Biol. 2008;129(6):667-686.
doi pubmed - Zhou J, Giannakakou P. Targeting microtubules for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2005;5(1):65-71.
doi pubmed - Sharma PS, Sharma R, Tyagi R. Inhibitors of cyclin dependent kinases: useful targets for cancer treatment. Curr Cancer Drug Targets. 2008;8(1):53-75.
doi pubmed - Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000-5005
doi pubmed - Warner SL, Stephens BJ, Von Hoff DD. Tubulin-associated proteins: Aurora and Polo-like kinases as therapeutic targets in cancer. Curr Oncol Rep. 2008;10(2):122-129.
doi pubmed - Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17(11):3052-3065.
doi pubmed - Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs. 2012;30(6):2411-2432.
doi pubmed - Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12(4):851-862.
doi - Hans F, Skoufias DA, Dimitrov S, Margolis RL. Molecular distinctions between Aurora A and B: a single residue change transforms Aurora A into correctly localized and functional Aurora B. Mol Biol Cell. 2009;20(15):3491-3502.
doi pubmed - Fancelli D, Berta D, Bindi S, Cameron A, Cappella P, Carpinelli P, Catana C, et al. Potent and selective Aurora inhibitors identified by the expansion of a novel scaffold for protein kinase inhibition. J Med Chem. 2005;48(8):3080-3084.
doi pubmed - Berdnik D, Knoblich JA. Drosophila Aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr Biol. 2002;12(8):640-647.
doi - Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13(8):691-697.
doi - Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18(1):51-63.
doi pubmed - Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20(2):189-193.
doi pubmed - Goepfert TM, Adigun YE, Zhong L, Gay J, Medina D, Brinkley WR. Centrosome amplification and overexpression of aurora A are early events in rat mammary carcinogenesis. Cancer Res. 2002;62(14):4115-4122.
pubmed - Nishida N, Nagasaka T, Kashiwagi K, Boland CR, Goel A. High copy amplification of the Aurora-A gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer Biol Ther. 2007;6(4):525-533.
doi pubmed - Jeng YM, Peng SY, Lin CY, Hsu HC. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res. 2004;10(6):2065-2071.
doi pubmed - Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, Shimomura K, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer. 2001;84(6):824-831.
doi pubmed - Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, Ward IM, et al. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet. 2005;37(4):401-406.
doi pubmed - Ouchi M, Fujiuchi N, Sasai K, Katayama H, Minamishima YA, Ongusaha PP, Deng C, et al. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem. 2004;279(19):19643-19648.
doi pubmed - Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36(1):55-62.
doi pubmed - Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, Cheng JQ. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279(50):52175-52182.
doi pubmed - Yang H, Ou CC, Feldman RI, Nicosia SV, Kruk PA, Cheng JQ. Aurora-A kinase regulates telomerase activity through c-Myc in human ovarian and breast epithelial cells. Cancer Res. 2004;64(2):463-467.
doi pubmed - Sun C, Chan F, Briassouli P, Linardopoulos S. Aurora kinase inhibition downregulates NF-kappaB and sensitises tumour cells to chemotherapeutic agents. Biochem Biophys Res Commun. 2007;352(1):220-225.
doi pubmed - Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4(11):842-854.
doi pubmed - Bolton MA, Lan W, Powers SE, McCleland ML, Kuang J, Stukenberg PT. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol Biol Cell. 2002;13(9):3064-3077.
doi pubmed - Welburn JP, Vleugel M, Liu D, Yates JR, 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38(3):383-392.
doi pubmed - Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 1998;17(3):667-676.
doi pubmed - Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10(12):825-841.
doi pubmed - Keen N, Taylor S. Mitotic drivers--inhibitors of the Aurora B Kinase. Cancer Metastasis Rev. 2009;28(1-2):185-195.
doi pubmed - Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002;21(4):483-492.
doi pubmed - Ota T, Suto S, Katayama H, Han ZB, Suzuki F, Maeda M, Tanino M, et al. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res. 2002;62(18):5168-5177.
pubmed - Sistayanarain A, Tsuneyama K, Zheng H, Takahashi H, Nomoto K, Cheng C, Murai Y, et al. Expression of Aurora-B kinase and phosphorylated histone H3 in hepatocellular carcinoma. Anticancer Res. 2006;26(5A):3585-3593.
pubmed - Tatsuka M, Katayama H, Ota T, Tanaka T, Odashima S, Suzuki F, Terada Y. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998;58(21):4811-4816.
pubmed - Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, Spalletti-Cernia D, et al. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab. 2005;90(2):928-935.
doi pubmed - Lin ZZ, Jeng YM, Hu FC, Pan HW, Tsao HW, Lai PL, Lee PH, et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer. 2010;10:461.
doi pubmed - Wu JC, Chen TY, Yu CT, Tsai SJ, Hsu JM, Tang MJ, Chou CK, et al. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem. 2005;280(10):9013-9022.
doi pubmed - Yasen M, Mizushima H, Mogushi K, Obulhasim G, Miyaguchi K, Inoue K, Nakahara I, et al. Expression of Aurora B and alternative variant forms in hepatocellular carcinoma and adjacent tissue. Cancer Sci. 2009;100(3):472-480.
doi pubmed - Bernard M, Sanseau P, Henry C, Couturier A, Prigent C. Cloning of STK13, a third human protein kinase related to Drosophila aurora and budding yeast Ipl1 that maps on chromosome 19q13.3-ter. Genomics. 1998;53(3):406-409.
doi pubmed - Yang KT, Li SK, Chang CC, Tang CJ, Lin YN, Lee SC, Tang TK. Aurora-C kinase deficiency causes cytokinesis failure in meiosis I and production of large polyploid oocytes in mice. Mol Biol Cell. 2010;21(14):2371-2383.
doi pubmed - Kimura M, Matsuda Y, Yoshioka T, Okano Y. Cell cycle-dependent expression and centrosome localization of a third human aurora/Ipl1-related protein kinase, AIK3. J Biol Chem. 1999;274(11):7334-7340.
doi pubmed - Brown JR, Koretke KK, Birkeland ML, Sanseau P, Patrick DR. Evolutionary relationships of Aurora kinases: implications for model organism studies and the development of anti-cancer drugs. BMC Evol Biol. 2004;4:39.
doi pubmed - Fernandez-Miranda G, Trakala M, Martin J, Escobar B, Gonzalez A, Ghyselinck NB, Ortega S, et al. Genetic disruption of aurora B uncovers an essential role for aurora C during early mammalian development. Development. 2011;138(13):2661-2672.
doi pubmed - Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10(3):262-267.
doi pubmed - Kitzen JJ, de Jonge MJ, Verweij J. Aurora kinase inhibitors. Crit Rev Oncol Hematol. 2010;73(2):99-110.
doi pubmed - Boss DS, Beijnen JH, Schellens JH. Clinical experience with aurora kinase inhibitors: a review. Oncologist. 2009;14(8):780-793.
doi pubmed - Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6(12 Pt 1):3158-3168.
doi pubmed - Benten D, Keller G, Quaas A, Schrader J, Gontarewicz A, Balabanov S, Braig M, et al. Aurora kinase inhibitor PHA-739358 suppresses growth of hepatocellular carcinoma in vitro and in a xenograft mouse model. Neoplasia. 2009;11(9):934-944.
doi pubmed - Fraedrich K, Schrader J, Ittrich H, Keller G, Gontarewicz A, Matzat V, Kromminga A, et al. Targeting aurora kinases with danusertib (PHA-739358) inhibits growth of liver metastases from gastroenteropancreatic neuroendocrine tumors in an orthotopic xenograft model. Clin Cancer Res. 2012;18(17):4621-4632.
doi pubmed - Cohen RB, Jones SF, Aggarwal C, von Mehren M, Cheng J, Spigel DR, Greco FA, et al. A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clin Cancer Res. 2009;15(21):6694-6701.
doi pubmed - Steeghs N, Eskens FA, Gelderblom H, Verweij J, Nortier JW, Ouwerkerk J, van Noort C, et al. Phase I pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol. 2009;27(30):5094-5101.
doi pubmed - Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, Dogliotti L, et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int. 2013;111(1):44-52.
doi pubmed - Ochiana SO, Pandarinath V, Wang Z, Kapoor R, Ondrechen MJ, Ruben L, Pollastri MP. The human Aurora kinase inhibitor danusertib is a lead compound for anti-trypanosomal drug discovery via target repurposing. Eur J Med Chem. 2013;62:777-784.
- Hajduch M, Vydra D, Dzubak P, et al. In vivo mode of action of CYC116, a novel small molecule inhibitor of aurora kinases and VEGFR2. Proc Am Cancer Res. 2008;49 abstr 5645.
- Griffiths G, Scaerou F, Midgley C, et al. Anti-tumour activity of CYC116, a novel small molecule inhibitor of aurora kinases and VEGFR2. Proc Am Assoc Cancer Res. 2008;49 abstr 5464.
- Maccallum D, Melville J, Cambell K, Green S. Combination studies with the oral aurora kinase inhibitor CYC116 and chemotherapeutic agents. Proc Am Assoc Cancer Res. 2008;49 abstr 4015.
- Leteur C, Tao Y, Deutch E, Bourhis J. Enhancement of radiation response by the aurora kinase inhibitor CYC116 in Ras mutant and p-53-deficient cancer cells. Proc Am Assoc cancer Res. 2009;50 abstr 2481.
- Arbitrario JP, Belmont BJ, Evanchik MJ, Flanagan WM, Fucini RV, Hansen SK, Harris SO, et al. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010;65(4):707-717.
doi pubmed - VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV. The Aurora kinase inhibitor SNS-314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule-targeted agents in a colon carcinoma model. Mol Cancer Ther. 2009;8(4):930-939.
doi pubmed - Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P, Ross S, et al. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010;70(23):9846-9854.
doi pubmed - Evans R, Naber C, Steffler T, Checkland T, Keats J, Maxwell C, Perry T, et al. Aurora A kinase RNAi and small molecule inhibition of Aurora kinases with VE-465 induce apoptotic death in multiple myeloma cells. Leuk Lymphoma. 2008;49(3):559-569.
doi pubmed - Lin ZZ, Hsu HC, Hsu CH, Yeh PY, Huang CY, Huang YF, Chen TJ, et al. The Aurora kinase inhibitor VE-465 has anticancer effects in pre-clinical studies of human hepatocellular carcinoma. J Hepatol. 2009;50(3):518-527.
doi pubmed - McLaughlin J, Markovtsov V, Li H, Wong S, Gelman M, Zhu Y, Franci C, et al. Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. J Cancer Res Clin Oncol. 2010;136(1):99-113.
doi pubmed - Ranshaw JS, Patnaik A, Gordon M, Beeram M, Fischer D, Gianella-Borradori A. A phase I two arm trial of AS703569 (R763) an orally available aurora kinase inhibitor in subjects with solid tumours. J Clin Oncol. 2007;25:18S.
- Jani JP, Arcari J, Bernardo V, Bhattacharya SK, Briere D, Cohen BD, Coleman K, et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol Cancer Ther. 2010;9(4):883-894.
doi pubmed - Schoffski P, Jones SF, Dumez H, Infante JR, Van Mieghem E, Fowst C, Gerletti P, et al. Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur J Cancer. 2011;47(15):2256-2264.
doi pubmed - Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, Devine LA, et al. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J Med Chem. 2009;52(2):379-388.
doi pubmed - Tanaka R, Squires MS, Kimura S, Yokota A, Nagao R, Yamauchi T, Takeuchi M, et al. Activity of the multitargeted kinase inhibitor, AT9283, in imatinib-resistant BCR-ABL-positive leukemic cells. Blood. 2010;116(12):2089-2095.
doi pubmed - Foran JM, Parandi F, O'Brien SM, Borthakur G, Rios M, Boone J, et al. A phase I and pharmacodynamic trial of AT9283 an aurora kinase inhibitor in patients with refractory leukaemia. ASCO Annual meeting 2008. Chicago, IL, USA, Abstract 2518.
- Anderson K, Lai Z, McDonald OB, Stuart JD, Nartey EN, Hardwicke MA, Newlander K, et al. Biochemical characterization of GSK1070916, a potent and selective inhibitor of Aurora B and Aurora C kinases with an extremely long residence time1. Biochem J. 2009;420(2):259-265.
doi pubmed - Hardwicke MA, Oleykowski CA, Plant R, Wang J, Liao Q, Moss K, Newlander K, et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol Cancer Ther. 2009;8(7):1808-1817.
doi pubmed - Hoar K, Chakravarty A, Rabino C, Wysong D, Bowman D, Roy N, Ecsedy JA. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27(12):4513-4525.
doi pubmed - Dees EC, Infante JR, Cohen RB, O'Neil BH, Jones S, von Mehren M, Danaee H, et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;67(4):945-954.
doi pubmed - Macarulla T, Cervantes A, Elez E, Rodriguez-Braun E, Baselga J, Rosello S, Sala G, et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol Cancer Ther. 2010;9(10):2844-2852.
doi pubmed - Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, Rosello S, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(17):4764-4774.
doi pubmed - Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, Manfredi M, et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012;18(17):4775-4784.
doi pubmed - Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17(24):7614-7624.
doi pubmed - Kelly KR, Ecsedy J, Medina E, Mahalingam D, Padmanabhan S, Nawrocki ST, Giles FJ, et al. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukaemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. 2011;15(10):2057-2070.
doi pubmed - Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer. 2010;55(1):26-34.
doi - Tung R, Kaul S, Diamond GA, Shah PK. Narrative review: drug-eluting stents for the management of restenosis: a critical appraisal of the evidence. Ann Intern Med. 2006;144(12):913-919.
doi pubmed - Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, Hanfstein B, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16(11):2190-2196.
doi pubmed - Okabe S, Tauchi T, Tanaka Y, Kimura S, Maekawa T, Ohyashiki K. Activity of histone deacetylase inhibitors and an Aurora kinase inhibitor in BCR-ABL-expressing leukemia cells: Combination of HDAC and Aurora inhibitors in BCR-ABL-expressing cells. Cancer Cell Int. 2013;13(1):32.
doi pubmed - Yen CC, Yeh CN, Cheng CT, Jung SM, Huang SC, Chang TW, Jan YY, et al. Integrating bioinformatics and clinicopathological research of gastrointestinal stromal tumors: identification of aurora kinase A as a poor risk marker. Ann Surg Oncol. 2012;19(11):3491-3499.
doi pubmed - Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, Neuville A, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
doi pubmed - Tatsuka M, Sato S, Kanda A, Miki T, Kamata N, Kitajima S, Kudo Y, et al. Oncogenic role of nuclear accumulated Aurora-A. Mol Carcinog. 2009;48(9):810-820.
doi pubmed - Reiter R, Gais P, Jutting U, Steuer-Vogt MK, Pickhard A, Bink K, Rauser S, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12(17):5136-5141.
doi pubmed - Zhang H, Chen X, Jin Y, Liu B, Zhou L. Overexpression of Aurora-A promotes laryngeal cancer progression by enhancing invasive ability and chromosomal instability. Eur Arch Otorhinolaryngol. 2012;269(2):607-614.
doi pubmed - Qi G, Ogawa I, Kudo Y, Miyauchi M, Siriwardena BS, Shimamoto F, Tatsuka M, et al. Aurora-B expression and its correlation with cell proliferation and metastasis in oral cancer. Virchows Arch. 2007;450(3):297-302.
doi pubmed - Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, Devarajan K, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3(140):ra67.
doi pubmed - Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, Baumgart A, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget. 2011;2(8):599-609.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.