










| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://www.jocmr.org |
Review
Volume 15, Number 6, June 2023, pages 283-291

Metabolomics in Acute Kidney Injury: The Experimental Perspective
Daniel Patschana, b, e ![]() , Susann Patschana, Igor Matyukhina, Meike Hoffmeisterb, c, Martin Lauxmannc, Oliver Rittera, b, Werner Dammermannb, d
, Susann Patschana, Igor Matyukhina, Meike Hoffmeisterb, c, Martin Lauxmannc, Oliver Rittera, b, Werner Dammermannb, d
aDepartment of Medicine 1, Cardiology, Angiology, Nephrology, University Hospital Brandenburg of the Brandenburg Medical School Theodor Fontane, Brandenburg, Germany
bFaculty of Health Sciences Brandenburg, Brandenburg Medical School Theodor Fontane, Germany
cInstitute of Biochemistry, Brandenburg Medical School Theodor Fontane, Brandenburg, Germany
dDepartment of Medicine 2, Gastroenterology, Diabetes, Endocrinology, University Hospital Brandenburg of the Brandenburg Medical School Theodor Fontane, Brandenburg, Germany
eCorresponding Author: Daniel Patschan, Department of Medicine 1, Cardiology, Angiology, University Hospital Brandenburg of the Brandenburg Medical School Theodor Fontane, 14770 Brandenburg, Germany
Manuscript submitted March 20, 2023, accepted May 8, 2023, published online June 29, 2023
Short title: Metabolomics in AKI
doi: https://doi.org/10.14740/jocmr4913
| Abstract | ▴Top |
Acute kidney injury (AKI) affects increasing numbers of in-hospital patients in Central Europe and the USA, the prognosis remains poor. Although substantial progress has been achieved in the identification of molecular/cellular processes that induce and perpetuate AKI, more integrated pathophysiological perspectives are missing. Metabolomics enables the identification of low-molecular-weight (< 1.5 kD) substances from biological specimens such as certain types of fluid or tissue. The aim of the article was to review the literature on metabolic profiling in experimental AKI and to answer the question if metabolomics allows the integration of distinct pathophysiological events such as tubulopathy and microvasculopathy in ischemic and toxic AKI. The following databases were searched for references: PubMed, Web of Science, Cochrane Library, Scopus. The period lasted from 1940 until 2022. The following terms were utilized: “acute kidney injury” OR “acute renal failure” OR “AKI” AND “metabolomics” OR “metabolic profiling” OR “omics” AND “ischemic” OR “toxic” OR “drug-induced” OR “sepsis” OR “LPS” OR “cisplatin” OR “cardiorenal” OR “CRS” AND “mouse” OR “mice” OR “murine” OR “rats” OR “rat”. Additional search terms were “cardiac surgery”, “cardiopulmonary bypass”, “pig”, “dog”, and “swine”. In total, 13 studies were identified. Five studies were related to ischemic, seven studies to toxic (lipopolysaccharide (LPS), cisplatin), and one study to heat shock-associated AKI. Only one study, related to cisplatin-induced AKI, was performed as a targeted analysis. The majority of the studies identified multiple metabolic deteriorations upon ischemia/the administration of LPS or cisplatin (e.g., amino acid, glucose, lipid metabolism). Particularly, abnormalities in the lipid homeostasis were shown under almost all experimental conditions. LPS-induced AKI most likely depends on the alterations in the tryptophan metabolism. Metabolomics studies provide a deeper understanding of pathophysiological links between distinct processes that are responsible for functional impairment/structural damage in ischemic or toxic or other types of AKI.
Keywords: AKI; Metabolomics; KRT; Recovery of kidney function; Survival
| Introduction | ▴Top |
Acute kidney injury (AKI): definition and epidemiology
AKI affects increasing numbers of hospitalized patients worldwide. It must be diagnosed if one of the following criteria are fulfilled: an increase in serum creatinine of at least 0.3 mg/dL, a 1.5-fold increase within 7 days, or a reduction in urine output to under 0.5 mL/kg/h for 6 h or longer [1]. These criteria have been documented in the 2012 revised “KDIGO clinical practice guidelines for acute kidney injury” [2]. In 2018, Hoste et al reported an average in-hospital AKI incidence of 15-30% [3]. In hospitalized subjects, the overall mortality of the syndrome ranges from 15% to 25% [3, 4]. The chance of survival is significantly lower in intensive care units, where up to 60% of all treated subjects acquire AKI. Under these circumstances, AKI has been identified as an independent predictor of death [5]. Certain AKI etiologies are associated with a disproportional increase in mortality risk. For instance, subjects with in-hospital-acquired cardiorenal syndrome type 3 (AKI induces secondary cardiac complications [6]) show an in-hospital chance of survival of only 50% [7]. The coincidence of hematooncological neoplasia, chemotherapy-induced sepsis, and kidney replacement therapy (KRT) requiring AKI has been associated with a mortality probability of 100% [8]. Also, individual AKI episodes have been shown to substantially reduce long-term (over years) survival [9], particularly in more severe cases (AKI “injury” or “failure” according to the RIFLE (risk, injury, failure, loss of kidney function and end-stage kidney disease) classification [10]). In addition, it can no longer be doubted that AKI is a potent risk factor for chronic kidney disease (CKD) [11]. Forni et al [12] documented a 105-fold hazard ratio increase for CKD, if AKI requires KRT.
AKI: etiology
The AKI etiology is quite heterogeneous [13]. Historically, three major AKI entities have been defined: pre-, intra-, and postrenal AKI. Postrenal AKI, the least frequent AKI entity (up to 10% [14]) potentially results from obstruction of the ureter or bladder or urethra. Many diseases may account for postrenal AKI such as urolithiasis, neoplasia, or neurological diseases that affect the activity of smooth muscle cells in the ureter or bladder. In a broader sense, “prerenal” encompasses all situations in which the effective perfusion of renal tissue is diminished (ischemia) [15]. The most common causes are fluid/blood loss, and acute heart failure. Another cause is cardiac surgery-associated AKI (CSA-AKI) (the second most common type of AKI after septic AKI in intensive care treated patients [16]). It results from various factors including nephrotoxins, ischemia/hypoxia, mechanical trauma, systemic inflammation, and cardiopulmonary bypass [17].
Kidney dysfunction is potentially reversible in the early stages of ischemia, prolonged hypoperfusion however increasingly induces structural damage such as the loss of tubular brush border, vacuolization, and tubular debris secondary to cell apoptosis/necrosis [18]. Prerenal AKI transits to intrarenal AKI. The so-called bilateral cortical necrosis is rarely diagnosed, occasionally in septic and postpartum AKI [19]. Nevertheless, tubular cell necrosis may also occur in patients with drug-induced AKI. The term “nephrotoxic” usually summarizes kidney-related side effects of various types of medications. Environmental contaminants may also act nephrotoxic (cadmium [20], mercury [21]), the overall clinical relevance is nevertheless lower. Regarding drug-induced AKI, the following mechanisms may be involved: tubular cell necrosis (aminoglycosides, vancomycin) [22, 23], interstitial nephritis (proton pump inhibitors) [24], renal vasoconstriction (amphotericin B) [22], tubular obstruction (sulfonamides) [25], and thrombotic microangiopathy (calcineurin inhibitors) [26]. Clinically, many patients acquire AKI for several reasons (e.g., sepsis with subsequent systemic vasodilation and septic cardiomyopathy, additional use of nephrotoxic drugs). Renal ischemia per se variably induces three pathophysiological processes or responses: tubulopathy, interstitial and systemic inflammation, and peritubular microvasculopathy [15, 27-29]. Microvascular damage is not restricted to peritubular capillaries but also affects glomerular function and structure [15]. Increased microvascular permeability causes interstitial edema in both the tubular and glomerular compartments. Glomerular/mesangial fluid accumulation potentially reduces the filtration rate even further.
In general, the clinical course of AKI subjects encompasses either complete or incomplete recovery of kidney function (ROKF) [30]. In some individuals, ROKF does not occur at all. If ROKF takes longer than 7 days after an acute event, AKI progresses to acute kidney disease (AKD) [31]. Respective individuals are at significantly higher risk of CKD in the long-term [11]. Also, the long-term survival probability decreases with increasing AKI severity. Thus, patients with so-called “fatal AKI (e.g., bilateral cortical necrosis [32]) show a 7-year survival probability that approximates the life expectancy of patients with end-stage kidney disease (ESKD) [9].
The “-omics”: concept
The cellular/molecular processes that induce and perpetuate kidney damage in ischemic and toxic AKI have intensively been studied in the past. However, more integrated approaches were missing over many years. In this regard, “-omics” studies potentially offer new perspectives. The overall aim of the so-called “-omics” concept is to identify cellular/tissue response patterns under both physiological and pathological conditions in a more integrated fashion. “-Omics” are datasets on the detection, quantification, and characterization of biological molecules. Omics studies allow large-scale analysis of numerous proteins, nucleic acids, or metabolites at the same time. Four major types of “-omics” have been established, genomics, transcriptomics, proteomics, and metabolomics [33]. Metabolomics encompasses the analysis of lower molecular weight (< 1.5 kD) substances in cells/certain types of tissue/biological fluids. The current review article discusses experimental data published so far. It particularly aimed to answer the question of whether metabolic profiling enables a more integrated approach to the complex pathophysiology of ischemic or toxic or other types of AKI.
We omit to review the history of metabolomics or the considerable number of methods [33-38] that have been established since the first description of metabolic profiling in 1948 [39]. Metabolic profiling is either being performed as untargeted or targeted analysis [33, 40]. According to Johnson et al [41], untargeted or global metabolomics allows the assessment of metabolites extracted from a biological sample, it can potentially reveal novel perturbations. Targeted metabolomics instead measures the concentrations of a predefined set of metabolites. Both approaches can be used for the generation of new hypotheses.
| Methods | ▴Top |
The following databases were searched for references: PubMed, Web of Science, Cochrane Library, and Scopus. The period lasted from 1940 until 2022. The following terms were utilized: “acute kidney injury” OR “acute renal failure” OR “AKI” AND “metabolomics” OR “metabolic profiling” OR “omics” AND “ischemic” OR “toxic” OR “drug-induced” OR “sepsis” OR “LPS” OR “cisplatin” OR “cardiorenal” OR “CRS” AND “mouse” OR “mice” OR “murine” OR “rats” OR “rat”. Additional search terms were “cardiac surgery”, “cardiopulmonary bypass”, “pig”, “dog”, and “swine”. The flow chart (Supplemental Material 1, www.jocmr.org) illustrates the searching procedure.
| Ischemic AKI | ▴Top |
The first study that needs to be discussed was published more than 10 years ago (Liu et al [42]). Sprague-Dawley rats were subjected to bilateral renal artery clamping for 45 min, either with or without L-carnitine pretreatment. The substance was used due to its known anti-oxidative properties [43]. Serum concentrations of creatinine and blood urea nitrogen (BUN) peaked in post-ischemic animals 24 h after reperfusion, an effect that was markedly abrogated by L-carnitine. So-called “high-performance liquid chromatography coupled with mass spectrometry” was employed for metabolic profiling, and analyses were performed from serum samples. Briefly, the concentrations of the following metabolites increased post-ischemia: lysophospholipids, free fatty acids, and nitrotyrosine. The activity of the enzyme phospholipase A2 and serum levels of malondialdehyde in contrast decreased. Finally, cortical superoxide dismutase activity was reduced. All effects were diminished in L-carnitine-treated mice. The authors proposed alterations of lipid metabolism as key events in ischemia/reperfusion-associated kidney damage.
Two years later (2014), Wei et al [44] published a manuscript on metabolomics in murine ischemic AKI; the aim was a “global analysis of the metabolic changes in renal IRI”. Bilateral renal ischemia was applied for 25 min, followed by reperfusion periods for either 2 or 48 h or 7 days. To achieve the goal, plasma and tissue samples from both the renal cortex and the medulla were analyzed with gas chromatography/mass spectrometry (GC/MS) and liquid chromatography/mass spectrometry (LC/MS). Renal ischemia significantly deteriorated kidney excretory function (serum creatinine and BUN peaked at 48 h, respectively), functional impairment was however (partly) reversible until day 7. Metabolomics identified 404 substances from tissue samples and 293 metabolites from plasma. Renal ischemia reduced the plasma availability of several substances in a significant manner: betaine, tyrosine, glutamine, proline, methionine, and others. Overall, the study revealed significant alterations of glucose, lipid, and purine metabolism. There was also evidence for affected osmotic regulation (mannitol, arabitol, threitol, pinitol). Finally, the tissue availability of certain prostaglandins was modulated. The reported findings were highly relevant since they indicated an affected energy supply (induction/perpetuation of tubular damage) and the stimulation of inflammatory processes (prostaglandin dysbalance). Thus, metabolomics truly helped to identify a “link” between hypoperfusion/ischemia and the hallmarks of ischemic AKI: tubulopathy, inflammation, and (micro)vasculopathy.
Huang et al [45] also investigated ischemic AKI but employed a unilateral approach. Only one renal artery was occluded (45 min, Fisher rats (F344)), the other organ remained perfused throughout. Reperfusion either lasted for 4 or 24 h. In addition to metabolomics, the authors also performed proteomics from cortical tissue samples. The proteomic analysis identified 363 out of 2,798 proteins with different expressions in post-ischemic as compared to contralateral kidneys. The differences were most prominent at 24 h. Ischemia particularly stimulated the synthesis of factors involved in stress signaling (heat shock protein 70 (HSP70) and heme-oxygenase 1 (HO-1)), coagulation, complement activity, and fatty acid metabolism [45]). For metabolic profiling, the authors used the following methods: nuclear magnetic resonance spectroscopy (NMR) or gas chromatography-mass spectrometry (GCxGC-MS). Metabolomics showed a substantial tissue increase of lipid metabolites such as palmitate, stearate, linoleate, 1-monopalmitin, cholesterol, and others. The findings already appeared 4 h after the ischemic insult. Intrarenal glucose levels were also diminished at hour 4. Another relevant observation was impaired mitochondrial function and thus adenosine triphosphate (ATP) production at 24 h. Comparable to the previous study, this investigation offers mechanistic “links” between ischemia and further consequences such as inflammation and tubular disfunction.
Fox et al [46] utilized an experimental model of cardiorenal syndrome type 3. The latter has been defined as a disorder in which various cardiac complications may arise in response to AKI [2]. The authors applied bilateral renal ischemia to 8 - 10 weeks old, male C57BL/6 mice for 22 min, respectively. Cardiac metabolic profiling was performed 4 and 24 h and 7 days later. Renal ischemia significantly diminished kidney excretory function, as reflected by elevated serum creatinine (24 h) and BUN (all times points). A total number of 124 metabolites was analyzed, and the concentrations of more than 40% of the substances were altered post-ischemia. Particularly, several amino acids were depleted, and oxidative stress was increased. Higher oxidative stress was also found in renal tissue samples. In addition, cardiac energy production processes were modulated, with stimulation of anaerobic ATP synthesis. In summary, the study confirmed experimental data from others, that showed a dysbalanced cardiac redox system after renal ischemia [47, 48]. It also confirmed the concept of cardio-renal or reno-cardiac cross-talk in AKI, and thus the concept of cardiorenal syndromes in general.
A more recent study from 2022 [49] addressed a very important topic in clinical nephrology: heart surgery-associated AKI. The authors utilized a piglet model of cardiopulmonary bypass including so-called deep hypothermic circulatory arrest (CPB/DHCA). Targeted metabolic profiling was performed from kidney tissue, urine, and serum samples. Ten out of 20 animals that received CPB/DHCA developed AKI during follow-up (4 h). Tissue analysis showed dysregulated tryptophan and purine metabolism. Urine analysis on the other hand revealed stimulation of anaerobic glycolysis. The metabolic patterns in tissue and urine samples did not resemble each other. Although the study additionally identified serum abnormalities (pyroglutamic acid - stress marker), the authors concluded that increased urinary anaerobic glycolysis may qualify as a tool for early AKI recognition in CPB.
| Toxic AKI | ▴Top |
In this paragraph, we also included lipopolysaccharide (LPS)-induced AKI models, which are commonly used to mimic microenvironmental conditions typically found in sepsis. Septic AKI on the other hand emerges due to various causes such as LPS exposure, renal malperfusion, and systemic hyperinflammation [50, 51].
The first study of interest was published in 2019 [52]. The authors employed a pig model of AKI, initiated by the administration of living Escherichia coli to induce sepsis. The utilization of larger vertebrate organisms (pigs as opposed to mice or rats) facilitated extensive hemodynamic monitoring including the following outcome variables: mean arterial blood pressure (MAP), systemic blood flow (QT), mean pulmonary arterial pressure, renal artery blood flow (QRA), and renal cortical blood flow (QRC). Sepsis was associated with lower QRA and QRC, the urine output decreased as well. Metabolic profiling revealed several abnormalities in kidney tissue, urine, and serum. Kidney tissue lactate and nicotinuric acid were elevated whereas the tissue concentrations of certain amino acids (e.g., valine, aspartate) and of glucose decreased. Serum analysis also revealed higher lactate levels, and glucose concentrations were diminished. Urine analysis showed higher levels of isovaleroglycine, aminoadipic acid, N-acetylglutamine, N-acetylaspartate, and ascorbic acid, myoinositol and phenylacetylglycine were lower in comparison to saline-treated controls. The concentrations of several metabolites in kidney tissue and urine significantly correlated with well-established AKI biomarkers such as neutrophile gelatinase-associated lipocalin (NGAL), leading to the conclusion that metabolomics enables the identification of novel AKI biomarker molecules.
A model of tubulotoxic AKI was studied by Qu et al [53]. Sprague-Dawley rats received intraperitoneal (i.p.) injections of cisplatin (either 7.5 mg/kg or 15 mg/kg), followed by urine and kidney tissue collection 72 h later. The authors used so-called high-performance liquid chromatography-time-of-flight mass spectrometry (HPLC-TOF/MS) and included 37 distinct metabolites in their analyses. Ultimately, seven major metabolic pathways were dysregulated (a detailed description shall be omitted), either involved in energy or amino acid, or lipid metabolism. The conclusion that respective abnormalities are partly responsible for increased oxidative stress and inflammation was legitimate. Additional urine analyses revealed four candidates as potential AKI biomarkers after cisplatin exposure. The study once more highlights one essential role of metabolomics in experimental and clinical AKI: the “screening” of yet unknown pathophysiological and diagnostic pathways/biomarkers.
An untargeted metabolomics approach was also favored by Gao et al [54]. The disease of interest was LPS-induced (multi)-organ failure. The authors used ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC/QTOF-MS) in rats that were intraperitoneally injected with LPS at 10 mg/kg once. Significant tissue damage was observed in the liver, lungs, colon, and kidney. Serum analysis showed alterations in a total number of 53 pathways with more than 120 aberrant metabolites (e.g., D-glutamine, D-glutamate, taurine, hypotaurine, and others). It was once more concluded that metabolic abnormalities potentially account for increased systemic inflammation and oxidative stress. The final sentence in the manuscript shall be cited: “The differential metabolites and metabolic pathways identified in this paper should be further studied using targeted metabolomics, lipidomics, and proteomics, in order to elucidate mechanisms and screening therapeutic targets for developing early diagnostic strategies and treatments.” The sentence somehow reflects the advantages but also the limitations of untargeted metabolomics in general. It potentially confirms or even modifies pathophysiological concepts of certain disease states such as AKI, but more specific conclusions often remain difficult.
This also applies to the next (untargeted) study, which was published in 2021 [55]. Once more, rats (Sprague-Dawley) underwent LPS treatment (i.p.), and analyses were performed either 2 or 6 h later. Thus, three groups (control (CT), LPS2, and LPS6) were defined, with significant increases in serum creatinine and BUN in the LPS6 group as compared to CT and LPS2 (P = 0.0009 and P = 0.001). In parallel, the kidney structure of both LPS2 and LPS6 mice was significantly affected (e.g., detachment of the brush border, epithelial cell shedding and, others). The detailed results will not be discussed, but the study ultimately revealed three key findings: LPS-induced stimulation of systemic aerobic and anaerobic metabolism, impaired oxygen supply, and abnormalities in fatty acid metabolism. These events were finally proposed as responsible for the development of sepsis-associated AKI.
A study on targeted metabolic profiling (liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS)) in cisplatin-induced AKI was published in 2021 [56]. The particular aim was to uncover cisplatin-induced alterations in the tryptophan metabolism. Previous studies identified the amino acid and its metabolites as potential biomarkers in CKD and AKI [57, 58]. Therefore, the study particularly focused on the following metabolites: tryptophan, 5-hydroxytryptamin (serotonin), N-acetylserotonin, 3-hydroxyanthranilic acid (HAA), kynurenine (KYN), indole-3-lactic acid (ILA), indole-3-acetamide (IAM), 5-methoxy-3-indoleacetic acid (MIAA), and others. Sprague-Dawley rats were intravenously injected with three different cisplatin doses (group “low” (L): 3.0 mg/kg; group “middle” (M): 6.0 mg/kg; group “high” (H): 9.0 mg/kg), and controls received saline only. Analyses of blood and renal tissue were performed on day 5 after drug exposure. Both analytes, serum creatinine, and BUN gradually increased from “L” to “M” to “H”. Tissue analysis was separately performed from cortical and medullary dissections, respectively. Cisplatin injection significantly affected the tryptophan metabolism in cortex and medulla, the medullary area however was more susceptible. Out of 29 studied metabolites, indoxyl sulfate accumulated in a dose-dependent manner. The functional relevance of indoxyl sulfate was proven by additional experiments with chlormethiazole, an inhibitor of CYP2E1 (a member of the cytochrome P450 mixed-function oxidase system). Reduced hepatic indoxyl sulfate synthesis attenuated cisplatin-induced AKI.
Several previously discussed studies identified AKI-associated dysregulation of lipid metabolism, not only in toxic but also in ischemic AKI. Xiong et al [59] performed their study in C57/BL6 mice and in Sprague-Dawley and Wistar rats. AKI was once more induced by i.p. injections of cisplatin (25 mg/kg); animals were euthanized 1, 2, and 4 days later. Metabolomics revealed an accumulation of triglycerides in renal tissue, and the findings were confirmed by oil red O staining. For a more detailed characterization of accumulated triglycerides, the authors used a mass spectrometry-based approach, therefore the investigation was ultimately a lipidomics study. Further analyses focused on the so-called superfamily of uncoupling proteins (UCPs), which are involved in lipid metabolism. The expression of UCPs 1 - 3 was evaluated in renal tissue, with strong signals for UCPs 1 and 2 under normal conditions (mice and rats). UCP 3 in contrast was hardly detectable at all. AKI induced a decrease in intrarenal UCP 1, and the loss was correlated with AKI severity. Adenovirus-based UCP 1 expression in kidneys of cisplatin-treated mice attenuated three outcome variables: serum creatinine decreased, tissue damage was reduced, and lipid accumulation was diminished. Finally, the authors showed a link between UCP 1 activity and the AMPK/ULK1/autophagy pathway. The study impressively showed impaired intrarenal lipid clearance, most likely resulting from reduced UCP 1 activity and impaired autophagy.
The central role of certain lipid components in the pathogenesis of cisplatin-induced AKI was also documented by Song et al [60]. The study aimed to identify the mechanisms by which the substance astragaloside IV (AS IV), an active compound of the traditional Chinese herb Astragalus membranaceus, may attenuate the effects of cisplatin.
| Other Types of AKI | ▴Top |
Xue et al [61] published an article on heat stroke (HS)-related AKI in 2021. HS potentially induces a systemic inflammatory response syndrome with or without AKI. According to Ren et al [62], two types of HS must be distinguished, exertional HS (EHS) and non-exertional HS (NEHS). HS mainly evolves in younger and physically active subjects, NEHS in contrast affects older individuals with a higher degree of cumulative morbidity. The authors established a murine HS model by increasing the animals’ body temperature to 41 °C. The final temperature was maintained only shortly, but serum creatinine significantly increased in HS as compared to control animals. Interestingly, HS mice showed an almost generalized distribution pattern of 18-fluorodeoxyglucose (18FDG) (analyzed by micro-positron emission tomography/computed tomography scanning). Immunoblot analysis revealed higher abundances of high-mobility-group-box protein 1 (HMGB1) and receptor for advanced glycation end products (RAGE) in renal tissue specimens from HS animals. Finally, liquid chromatography-mass spectrometry showed an enrichment of unsaturated fatty acids. The authors concluded a key role for HMGB1/RAGE and unsaturated fatty acids in AKI induction post-HS.
Figure 1 summarizes metabolomics-derived findings in experimental AKI. The figure hardly differentiates between specific areas within the kidney but is intended to illustrate metabolic alterations that occur in ischemic/toxic/other types of AKI in a more general manner.
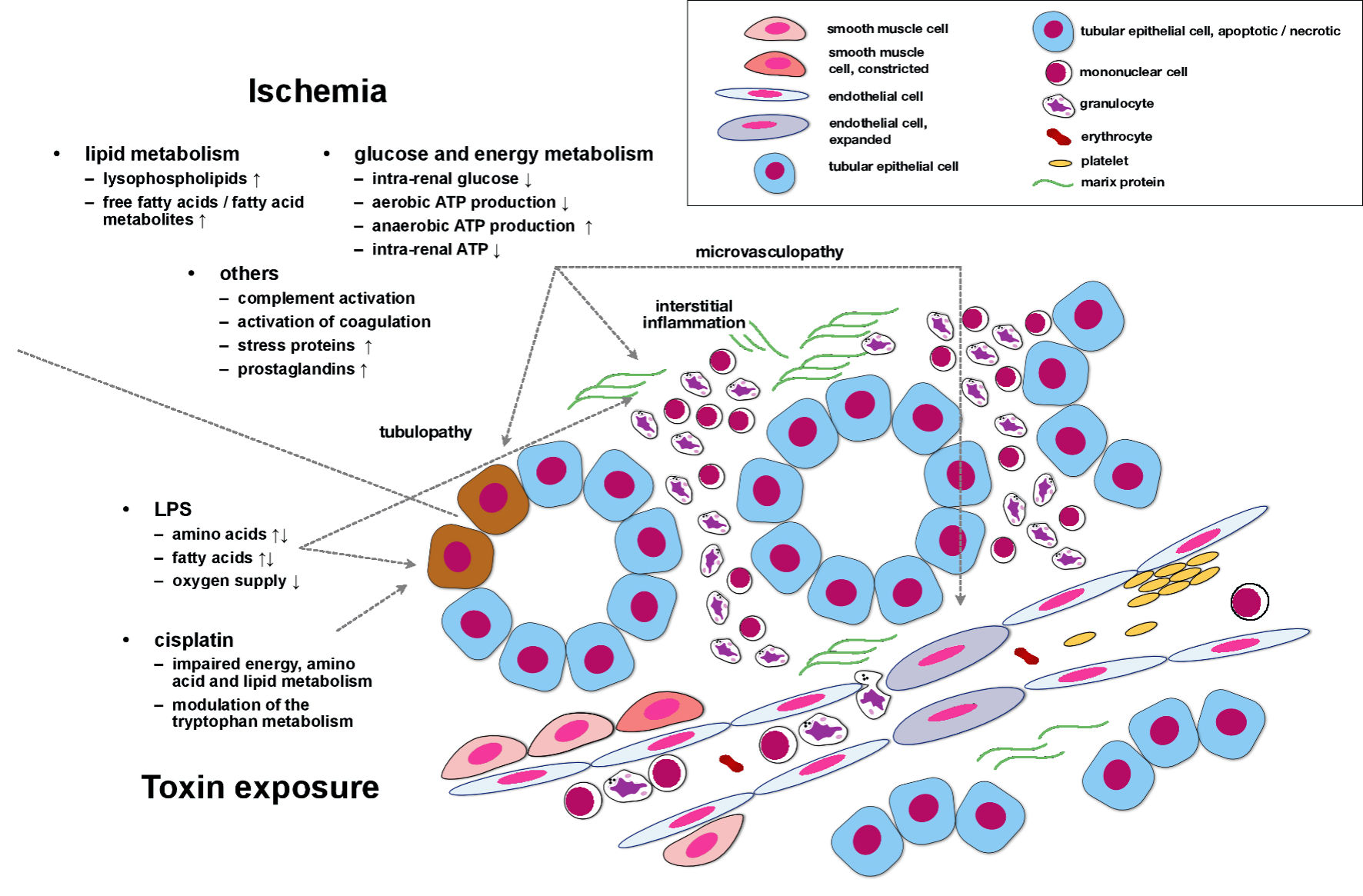 Click for large image | Figure 1. Metabolomics-derived findings in experimental AKI. Ischemia induces multiple deteriorations of lipid, glucose, and energy metabolism. In addition, the complement activity increases, coagulation pathways are activated, and prostaglandin synthesis is stimulated. These events, in conjunction, induce and perpetuate tubular cell dysfunction/damage, interstitial inflammation, and microvasculopathy. Under experimental conditions, both LPS and cisplatin also impair lipid and energy metabolism. Cisplatin particularly modulates the tryptophan pathway, most likely a key mechanism in cisplatin-induced AKI. LPS: lipopolysaccharide; AKI: acute kidney injury; ATP: adenosine triphosphate. |
Table 1 [42, 44-46, 49, 52-56, 59, 61] summarizes all studies discussed in the text, including reference, year, design, and essential findings.
 Click to view | Table 1. Summary of All Experimental Studies Discussed in the Text |
| Conclusions | ▴Top |
Will metabolomics studies provide a deeper understanding of pathophysiological processes that are responsible for functional impairment/structural damage in ischemic or toxic or other types of AKI? In our opinion, metabolic profiling offers valuable information in this respect. Several studies identified abnormalities in amino acid, glucose, and lipid metabolism. Particularly the role of the latter may not be underestimated, as shown by the study of Xiong et al [59]. One investigation even found impaired energy and prostaglandin metabolism [44] in response to ischemia, both in conjunction, potentially induce tubular dysfunction/damage and stimulated inflammation.
| Supplementary Material | ▴Top |
Suppl 1. Flow chart: the searching procedure.
Acknowledgments
None to declare.
Financial Disclosure
No funding was provided for the study.
Conflict of Interest
The authors declare that they do not have any conflict of interest.
Author Contributions
Daniel Patschan and Susann Patschan wrote the article. Igor Matyukhin searched for references. Meike Hoffmeister and Martin Lauxmann searched for references and assisted in figure preparation. Oliver Ritter provided financial support and assisted in writing. Werner Dammermann designed the manuscript and helped in writing. All authors finally approved the final version of the manuscript.
Data Availability
All data discussed in the article were extracted from the references cited in the text and the bibliography.
| References | ▴Top |
- Mercado MG, Smith DK, Guard EL. Acute kidney injury: diagnosis and management. Am Fam Physician. 2019;100(11):687-694.
pubmed - Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120(4):c179-184.
doi pubmed - Hoste EAJ, Kellum JA, Selby NM, Zarbock A, Palevsky PM, Bagshaw SM, Goldstein SL, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol. 2018;14(10):607-625.
doi pubmed - Asmus K, Erfurt S, Ritter O, Patschan S, Patschan D. AKI epidemiology and outcomes: a retrospective cohort study from the prenephrology era. Int J Nephrol. 2021;2021:5549316.
doi pubmed pmc - Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL, et al. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol. 2013;8(9):1482-1493.
doi pubmed pmc - Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52(19):1527-1539.
doi pubmed - Drubel K, Marahrens B, Ritter O, Patschan D. Kidney-related outcome in cardiorenal syndrome type 3. Int J Nephrol. 2022;2022:4895434.
doi pubmed pmc - Heeg M, Mertens A, Ellenberger D, Muller GA, Patschan D. Prognosis of AKI in malignant diseases with and without sepsis. BMC Anesthesiol. 2013;13(1):36.
doi pubmed pmc - Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol. 2014;10(4):193-207.
doi pubmed - Bagshaw SM, George C, Bellomo R, Committe ADM. A comparison of the RIFLE and AKIN criteria for acute kidney injury in critically ill patients. Nephrol Dial Transplant. 2008;23(5):1569-1574.
doi pubmed - Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52.
doi pubmed - Forni LG, Darmon M, Ostermann M, Oudemans-van Straaten HM, Pettila V, Prowle JR, Schetz M, et al. Renal recovery after acute kidney injury. Intensive Care Med. 2017;43(6):855-866.
doi pubmed pmc - Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. 2019;394(10212):1949-1964.
doi pubmed - Goyal A, Daneshpajouhnejad P, Hashmi MF, Bashir K. Acute Kidney Injury. In: StatPearls. Treasure Island (FL). 2023.
pubmed pmc - Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2(2):1303-1353.
doi pubmed pmc - Wang Y, Bellomo R. Cardiac surgery-associated acute kidney injury: risk factors, pathophysiology and treatment. Nat Rev Nephrol. 2017;13(11):697-711.
doi pubmed - Thiele RH, Isbell JM, Rosner MH. AKI associated with cardiac surgery. Clin J Am Soc Nephrol. 2015;10(3):500-514.
doi pubmed pmc - Patschan D, Hildebrandt A, Rinneburger J, Wessels JT, Patschan S, Becker JU, Henze E, et al. The hormone melatonin stimulates renoprotective effects of "early outgrowth" endothelial progenitor cells in acute ischemic kidney injury. Am J Physiol Renal Physiol. 2012;302(10):F1305-1312.
doi pubmed - Nwoko R, Plecas D, Garovic VD. Acute kidney injury in the pregnant patient. Clin Nephrol. 2012;78(6):478-486.
doi pubmed - Gong ZG, Zhao Y, Wang ZY, Fan RF, Liu ZP, Wang L. Epigenetic regulator BRD4 is involved in cadmium-induced acute kidney injury via contributing to lysosomal dysfunction, autophagy blockade and oxidative stress. J Hazard Mater. 2022;423(Pt A):127110.
doi pubmed - Rojas-Franco P, Franco-Colin M, Torres-Manzo AP, Blas-Valdivia V, Thompson-Bonilla MDR, Kandir S, Cano-Europa E. Endoplasmic reticulum stress participates in the pathophysiology of mercury-caused acute kidney injury. Ren Fail. 2019;41(1):1001-1010.
doi pubmed pmc - Pierson-Marchandise M, Gras V, Moragny J, Micallef J, Gaboriau L, Picard S, Choukroun G, et al. The drugs that mostly frequently induce acute kidney injury: a case - noncase study of a pharmacovigilance database. Br J Clin Pharmacol. 2017;83(6):1341-1349.
doi pubmed pmc - Bamgbola O. Review of vancomycin-induced renal toxicity: an update. Ther Adv Endocrinol Metab. 2016;7(3):136-147.
doi pubmed pmc - Antoniou T, Macdonald EM, Hollands S, Gomes T, Mamdani MM, Garg AX, Paterson JM, et al. Proton pump inhibitors and the risk of acute kidney injury in older patients: a population-based cohort study. CMAJ Open. 2015;3(2):E166-171.
doi pubmed pmc - Schwarz A, Perez-Canto A. Nephrotoxicity of antiinfective drugs. Int J Clin Pharmacol Ther. 1998;36(3):164-167.
pubmed - Choi CM, Schmaier AH, Snell MR, Lazarus HM. Thrombotic microangiopathy in haematopoietic stem cell transplantation: diagnosis and treatment. Drugs. 2009;69(2):183-198.
doi pubmed - Patschan D, Kribben A, Muller GA. Postischemic microvasculopathy and endothelial progenitor cell-based therapy in ischemic AKI: update and perspectives. Am J Physiol Renal Physiol. 2016;311(2):F382-394.
doi pubmed - Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281(5):F887-899.
doi pubmed - Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17(6):1503-1520.
doi pubmed - Pannu N, James M, Hemmelgarn B, Klarenbach S, Alberta Kidney Disease N. Association between AKI, recovery of renal function, and long-term outcomes after hospital discharge. Clin J Am Soc Nephrol. 2013;8(2):194-202.
doi pubmed pmc - Chawla LS, Bellomo R, Bihorac A, Goldstein SL, Siew ED, Bagshaw SM, Bittleman D, et al. Acute kidney disease and renal recovery: consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat Rev Nephrol. 2017;13(4):241-257.
doi pubmed - Frimat M, Decambron M, Lebas C, Moktefi A, Lemaitre L, Gnemmi V, Sautenet B, et al. Renal cortical necrosis in postpartum hemorrhage: a case series. Am J Kidney Dis. 2016;68(1):50-57.
doi pubmed - Dai X, Shen L. Advances and Trends in Omics Technology Development. Front Med (Lausanne). 2022;9:911861.
doi pubmed pmc - Jurowski K, Kochan K, Walczak J, Baranska M, Piekoszewski W, Buszewski B. Analytical techniques in lipidomics: state of the art. Crit Rev Anal Chem. 2017;47(5):418-437.
doi pubmed - Jurczak E, Mazurek AH, Szeleszczuk L, Pisklak DM, Zielinska-Pisklak M. Pharmaceutical hydrates analysis-overview of methods and recent advances. Pharmaceutics. 2020;12(10):E959.
doi pubmed pmc - Eberhardt K, Stiebing C, Matthaus C, Schmitt M, Popp J. Advantages and limitations of Raman spectroscopy for molecular diagnostics: an update. Expert Rev Mol Diagn. 2015;15(6):773-787.
doi pubmed - Eom JS, Kim ET, Kim HS, Choi YY, Lee SJ, Lee SS, Kim SH, et al. Metabolomics comparison of rumen fluid and milk in dairy cattle using proton nuclear magnetic resonance spectroscopy. Anim Biosci. 2021;34(2):213-222.
doi pubmed pmc - Sun J, Cao Z, Schnackenberg L, Pence L, Yu LR, Choudhury D, Palevsky PM, et al. Serum metabolite profiles predict outcomes in critically ill patients receiving renal replacement therapy. J Chromatogr B Analyt Technol Biomed Life Sci. 2021;1187:123024.
doi pubmed - Williams RJ, Kirby H. Paper chromatography using capillary ascent. Science. 1948;107(2784):481-483.
doi pubmed - Kim SJ, Song HE, Lee HY, Yoo HJ. Mass spectrometry-based metabolomics in translational research. Adv Exp Med Biol. 2021;1310:509-531.
doi pubmed - Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17(7):451-459.
doi pubmed pmc - Liu Y, Yan S, Ji C, Dai W, Hu W, Zhang W, Mei C. Metabolomic changes and protective effect of (L)-carnitine in rat kidney ischemia/reperfusion injury. Kidney Blood Press Res. 2012;35(5):373-381.
doi pubmed - Ergun O, Ulman I. Protective effect of L-carnitine on renal ischaemia-reperfusion injury in the rat. Cell Biochem Funct. 2005;23(5):369.
doi pubmed - Wei Q, Xiao X, Fogle P, Dong Z. Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS One. 2014;9(9):e106647.
doi pubmed pmc - Huang H, van Dullemen LFA, Akhtar MZ, Faro ML, Yu Z, Valli A, Dona A, et al. Proteo-metabolomics reveals compensation between ischemic and non-injured contralateral kidneys after reperfusion. Sci Rep. 2018;8(1):8539.
doi pubmed pmc - Fox BM, Gil HW, Kirkbride-Romeo L, Bagchi RA, Wennersten SA, Haefner KR, Skrypnyk NI, et al. Metabolomics assessment reveals oxidative stress and altered energy production in the heart after ischemic acute kidney injury in mice. Kidney Int. 2019;95(3):590-610.
doi pubmed pmc - Caio-Silva W, da Silva Dias D, Junho CVC, Panico K, Neres-Santos RS, Pelegrino MT, Pieretti JC, et al. Characterization of the oxidative stress in renal ischemia/reperfusion-induced cardiorenal syndrome type 3. Biomed Res Int. 2020;2020:1605358.
doi pubmed pmc - Patschan D, Marahrens B, Jansch M, Patschan S, Ritter O. Experimental cardiorenal syndrome type 3: what is known so far? J Clin Med Res. 2022;14(1):22-27.
doi pubmed pmc - Davidson JA, Robison J, Khailova L, Frank BS, Jaggers J, Ing RJ, Lawson S, et al. Metabolomic profiling demonstrates evidence for kidney and urine metabolic dysregulation in a piglet model of cardiac surgery-induced acute kidney injury. Am J Physiol Renal Physiol. 2022;323(1):F20-F32.
doi pubmed pmc - Bellomo R, Kellum JA, Ronco C, Wald R, Martensson J, Maiden M, Bagshaw SM, et al. Acute kidney injury in sepsis. Intensive Care Med. 2017;43(6):816-828.
doi pubmed - Gomez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016;22(6):546-553.
doi pubmed pmc - Izquierdo-Garcia JL, Nin N, Cardinal-Fernandez P, Rojas Y, de Paula M, Granados R, Martinez-Caro L, et al. Identification of novel metabolomic biomarkers in an experimental model of septic acute kidney injury. Am J Physiol Renal Physiol. 2019;316(1):F54-F62.
doi pubmed - Qu X, Gao H, Sun J, Tao L, Zhang Y, Zhai J, Song Y, et al. Identification of key metabolites during cisplatin-induced acute kidney injury using an HPLC-TOF/MS-based non-targeted urine and kidney metabolomics approach in rats. Toxicology. 2020;431:152366.
doi pubmed - Gao H, Yang T, Chen X, Song Y. Changes of lipopolysaccharide-induced acute kidney and liver injuries in rats based on metabolomics analysis. J Inflamm Res. 2021;14:1807-1825.
doi pubmed pmc - Ping F, Li Y, Cao Y, Shang J, Zhang Z, Yuan Z, Wang W, et al. Metabolomics analysis of the development of sepsis and potential biomarkers of sepsis-induced acute kidney injury. Oxid Med Cell Longev. 2021;2021:6628847.
doi pubmed pmc - Tan B, Chen J, Qin S, Liao C, Zhang Y, Wang D, Li S, et al. Tryptophan pathway-targeted metabolomics study on the mechanism and intervention of cisplatin-induced acute kidney injury in rats. Chem Res Toxicol. 2021;34(7):1759-1768.
doi pubmed - Li Y, Zhang X, Zhou H, Fan S, Wang Y, Zhang L. Metabonomics study on nephrotoxicity induced by intraperitoneal and intravenous cisplatin administration using rapid resolution liquid chromatography coupled with quadrupole-time-of-flight mass spectrometry (RRLC-Q-TOF-MS). RSC Adv. 2014;4(16):8260-8270.
- Kim HS, Kundu A, Son JY. Identification of sensitive biomarker, 3-indoxyl sulfate, to detecting acute kidney injury. Toxicol Lett. 2017;280:S238
- Xiong W, Xiong Z, Song A, Lei C, Ye C, Zhang C. Relieving lipid accumulation through UCP1 suppresses the progression of acute kidney injury by promoting the AMPK/ULK1/autophagy pathway. Theranostics. 2021;11(10):4637-4654.
doi pubmed pmc - Song Y, Hu T, Gao H, Zhai J, Gong J, Zhang Y, Tao L, et al. Altered metabolic profiles and biomarkers associated with astragaloside IV-mediated protection against cisplatin-induced acute kidney injury in rats: An HPLC-TOF/MS-based untargeted metabolomics study. Biochem Pharmacol. 2021;183:114299.
doi pubmed - Xue L, Guo W, Li L, Ou S, Zhu T, Cai L, Ding W, et al. Metabolomic profiling identifies a novel mechanism for heat stroke-related acute kidney injury. Mol Med Rep. 2021;23(4):241.
doi pubmed pmc - Ren MQ, Kazman JB, Abraham PA, Atias-Varon D, Heled Y, Deuster PA. Gene expression profiling of humans under exertional heat stress: Comparisons between persons with and without exertional heat stroke. J Therm Biol. 2019;85:102423.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.