










| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 9, Number 4, April 2017, pages 317-331
Molecular Analysis of Factor VIII and Factor IX Genes in Hemophilia Patients: Identification of Novel Mutations and Molecular Dynamics Studies
Faisal A. Al-Allafa, b, c, e, f, Mohiuddin M. Tahera, c, e, f, Zainularifeen Abduljaleela, c, Abdellatif Bouazzaouia, c, Mohammed Athara, c, Neda M. Bogaria, Halah A. Abalkhaild, Tarek MA. Owaidahd
aFaculty of Medicine, Department of Medical Genetics, Umm Al-Qura University, Makkah, Kingdom of Saudi Arabia
bMolecular Diagnostics Unit, Department of Laboratory Medicine and Blood Bank, King Abdullah Medical City, Makkah, Kingdom of Saudi Arabia
cScience and Technology Unit, Umm Al-Qura University, Makkah, Kingdom of Saudi Arabia
dPathology and Laboratory Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Kingdom of Saudi Arabia
eThese authors contributed equally to this study.
fCorresponding Authors: Mohiuddin M. Taher, Faculty of Medicine, Department of Medical Genetics, Umm Al-Qura University, Makkah, Kingdom of Saudi Arabia; Faisal A. Al-Allaf, Faculty of Medicine, Department of Medical Genetics, Umm Al-Qura University, Makkah, Kingdom of Saudi Arabia
Manuscript accepted for publication January 23, 2017
Short title: FVIII and FIX Mutations in Saudi Patients
doi: https://doi.org/10.14740/jocmr2876w
| Abstract | ▴Top |
Background: Hemophilias A and B are X-linked bleeding disorders caused by mutations in the factor VIII and factor IX genes, respectively. Our objective was to identify the spectrum of mutations of the factor VIII and factor IX genes in Saudi Arabian population and determine the genotype and phenotype correlations by molecular dynamics (MD) simulation.
Methods: For genotyping, blood samples from Saudi Arabian patients were collected, and the genomic DNA was amplified, and then sequenced by Sanger method. For molecular simulations, we have used softwares such as CHARMM (Chemistry at Harvard Macromolecular Mechanics; http://www.charmm-gui.org) and GROMACS. In addition, the secondary structure was determined based on the solvent accessibility for the confirmation of the protein stability at the site of mutation.
Results: Six mutations (three novel and three known) were identified in factor VIII gene, and six mutations (one novel and five known) were identified in factor IX gene. The factor VIII novel mutations identified were c.99G>T, p. (W33C) in exon 1, c.2138 DelA, p. (N713Tfs*9) in eon14, also a novel mutation at splicing acceptor site of exon 23 c.6430 - 1G>A. In factor IX, we found a novel mutation c.855G>C, p. (E285D) in exon 8. These novel mutations were not reported in any factor VIII or factor IX databases previously. The deleterious effects of these novel mutations were confirmed by PolyPhen2 and SIFT programs.
Conclusion: The protein functional and structural studies and the models built in this work would be appropriate for predicting the effects of deleterious amino acid substitutions causing these genetic disorders. These findings are useful for genetic counseling in the case of consanguineous marriages which is more common in the Saudi Arabia.
Keywords: Hemophilia A; Hemophilia B; Saudi Arabia; Blood coagulation; Computer simulation
| Introduction | ▴Top |
Hemophilias are disorders of blood coagulation factors deficiencies [1, 2], and hemophilic patients suffer from prolonged bleeding time after trauma or injuries [3]. Hemophilia A (OMIM 306700) and hemophilia B (OMIM 306900) are sex-linked genetic disorders resulting in deficiency of plasma coagulant activities of factor VIII and factor IX, respectively [4, 5]. Both factor VIII and factor IX genes are located on the X chromosome which presents as two copies in females (XX); however, males have only one (XY) copy. Therefore, a female with a mutation in one copy will not develop the disease, but becomes a carrier of this trait since the other copy of the gene compensates with a normal product (factor VIII or factor IX). In some females, though it occurs rarely, presence of two defective copies results in hemophilia disorder [6]. On the other hand, males are more affected than females because they have only one copy of the X chromosome. Male patients do not transmit the disease to their sons, as they are not responsible for the X chromosome transmission, but rather the Y chromosome (responsible for male characteristics), but all their daughters will be carriers [7].
Hemophilia affects people from all racial and ethnic groups [3, 7]. Hemophilia A and hemophilia B are the most common, over 80% of patients have hemophilia A, and it is about four times as common as hemophilia B, and about half of those affected have the severe form [7]. Other coagulation factor deficiencies are less common, with patients suffering either milder bleeding or thrombotic episodes. The prevalence of hemophilia B is 1 in 30,000 males, whereas hemophilia A affects 1 in 5,000 males [5]. A deficiency in factor VIII or factor IX plasma levels leads to this coagulopathy, the primary causative defect in hemophilia A and B patients is mutations or deletions in the blood coagulation factors VIII and IX [1, 8 , 9].
Factor VIII and factor IX proteins are synthesized in the liver. Factor IX is synthesised as 461 amino acid residues, including signal peptide of 46 amino acids at its N-terminus [10]. Factor IX contains eight exons, whereas factor VIII gene is composed of 26 exons and encodes a mature protein of 2,332 amino acids [1, 8, 11]. Both proteins circulate in the blood as single-chain glycoproteins of inactive zymogens [11, 12]. Factor VIII mainly circulates in a stable complex with von Willebrand factor, it consists of six domains: A1-A2-B-A3-C1-C2, and is homologous to factor V. Thrombin activates the factor VIII by proteolytic cleavage of the B domain. Upon activation, factor VIIIa dissociates from the complex to interact with factor IXa in the coagulation cascade. It is a cofactor to factor IXa in the activation of factor X, which, in turn, with its cofactor factor Va, activates more thrombin. Then, fibrinogen is cleaved by thrombin into fibrin which polymerizes and crosslinks (using factor XIII) into a blood clot. When coagulation is initiated, factor IX is converted to an active form (FIXa) by proteolytic cleavage, resulting in an N-terminal light chain and a C-terminal heavy chain held together by one or more disulfide bonds [9]. The role of FIXa in the blood coagulation cascade is to activate factor X through interactions with calcium ions, membrane phospholipids, and factor VIII. Severity of hemophilias is associated with the type and position of the mutations in factor VIII and factor IX genes [5]. In normal conditions, hematological ranges for factor VIII and factor IX are 0.5 - 2 and 0.7 - 1.2 IU/mL, respectively [13]. The clinical manifestations of hemophilias A and B are indistinguishable; however, depending on factor VIII or factor IX plasma levels, hemophilias are classified [7, 14] as severe < 0.01 IU/mL (<1%), moderate 0.01 - 0.05 IU/mL (1-5%), and mild 0.05 - 0.4 IU/mL (5-30%).
Hemophilia care in Western and European countries is a well-organized system providing quality services to all hemophiliacs, through the hemophilia treatment centers [14, 15]; however, such an organized care is lacking in Arab countries. We estimated that at least around 1,000 Saudis are most likely affected by hemophilias A and B. The guidelines for treatment and management of hemophilias are published for Saudi Arabia by the Ministry of Health (MOH.Gov.Sa) and for Gulf countries (GCC) [16, 17], and recently, a national registry was established to collect the data of hemophilia patients at King Faisal Specialist Hospital and Research Centre (KFSH&RC). There are currently 2,015 unique variants in the factor VIII gene (factorviii-db.org). In recent years, construction of mutation databases for various diseases became popular, due to computer technologies, and for hemophilia, also several databases are available; however, contributions for these databases are very rare from the Arab countries [18, 19]. Unfortunately, there are not many reports establishing nature of common mutations causing hemophilia A or B in Saudi Arabian population. Only there were three reports on factor VIII mutations findings [20-22]. In this study, we plan to investigate the mutations in factor VIII and factor IX genes from Saudi hemophilia patients and we want to develop a genetic testing based on PCR and DNA sequencing. These tests will offer a fast and reliable screening method for carrier testing and prenatal genetic diagnosis to families at risk of having a child with hemophilia.
| Materials and Methods | ▴Top |
Patient’s data and DNA analysis
For factor VIII and for factor IX investigations, samples from Saudi Arabian families undergoing treatment at KFSH&RC were collected. All patients were tested for factor VIII and factor IX coagulant activities that were carried out on Behring Coagulation System (BCS; Siemens, Marburg GmbH, Germany). The study approval was obtained from the ethical committee (IRB) of Umm Al-Qura University, Makkah, and the IRB committee of KFSH&RC. British Committee for Standards in Haematology Guidelines (bcshguidelines.org) was followed for the patient’s selection criteria. Consent to participation in this was obtained from all patients or legal guardians for minors. Initially factor VIII DNA samples were screened for inversion 22 using KAPA2G Robust HotStart ready mix (cat. #KK5702) by inverse PCR as reported [20]. The DNA samples negative for inversion 22 were screened for inversion 1 by multiplexed PCR using Fermentas kit (cat. #K1082) as reported by us [20]. The cases negative for both inversions were used for capillary sequencing. Descriptions of the primers used for PCR amplification were reported previously for factor VIII [23, 24] and for factor IX [25]. PCR was performed using a HotStarTaq Plus DNA Polymerase Kit (Qiagen), followed by capillary electrophoresis as reported [20]. PCR conditions were as follows: Taq polymerase was activated at 94 °C for 5 min, followed by 35 cycles of denaturing at 94 °C for 30 s, annealing at 60 °C for 30 s, extension at 68 °C for 60 s, and final extension at 68 °C for 5 min. Capillary sequencing (Sanger method) with Big Dye 3.1 reaction was performed on ABI 3500 Genetic analyzer as described by Al-Allaf et al [20] for factor VIII all 26 exons, and as described by Mahajan et al (2004) for all eight exons of factor IX gene [25]. Nucleotide numbering is for coding bases from A (nucleotide +1) the initiation methionine (ATG) at position -171 (factor VIII mRNA gene bank ref. NM_000132) and (ATG) at position -29 (factor IX mRNA gene bank ref. NM_000133), respectively. The final mutation analysis was performed on CLC genomics workbench, and mutations were designated according to the Human Genome Variation Society (HGVS) guidelines [26].
Functional analysis using molecular dynamics (MD) simulation, protein conformational changes, stability, and solvent accessibility
In order to predict the possible impact of a variation on the function of factor VIII and factor IX genes, the online tools such as sorting intolerant from tolerant (SIFT) and polymorphism phenotyping (PolyPhen2) were used [27, 28]. For molecular simulations of factor VIII protein, we have used the commonly used softwares such as CHARMM (Chemistry at Harvard Macromolecular Mechanics; http://www.charmm-gui.org) and GROMACS v4.0.527. The MD simulations were performed with a 2 fs time step using a set temperature of 300 K and a constant pressure of 1 atm, below periodic solvent boundary conditions, with macromolecular mechanics based on atomic coordinates and dynamics trajectories. The knowledge-based minimization (KoBaMIN) program allowed refining the protein domain structures to evaluate changes as a result of refined protocol [29]. These simulations normally follow energy minimization that allows optimization of the protein structure and simulate its natural motion. The conjugate gradient technique for 3D structure optimization along with the deviation between the structures was assessed by their root mean square deviation (RMSD) values. This server utilizes a conjugate gradient algorithm for energy minimization for force field energy minimization and limited reminiscence Broyden-Fletcher-Goldfarb-Shanno (L-BFGS) methods [30]. The free energy simulations were performed with solvent water molecules in close proximity to the solute to get an efficient solvent boundary potential (SSBP). The configuration of ions was first verified by brief Monte-Carlo (MC) simulation based on van der Waals (VdW) interactions. The particle mesh Ewald methodology was utilized for electrostatics observations and a 12 Å cut-off was applied for VdW interactions [31]. The mutation was further studied utilizing (collaborative computational project, number 4 (CCP4); QtMG and energy minimization for 3D structures were carried out using NOMAD-Ref web server. The KCl was included to neutralize entire negative charge of the structures. Molecular operation environment (MOE) was used to assess the associated hemophilia A disease variants in the factor VIII protein. The whole simulation of the protein in water was performed to acquire molecular structures by immersing it in a solvent based on simulation parameters, and energy minimization to analyze potential energies, structural fluctuations, coordinate stability and geometrical features. The energy minimization of the mutant protein structure was executed by way of atomic non-local environment assessment (ANOLEA), a server that carries out energy calculations of a protein sequence and investigates the “non-local environment” (NLE) of every heavy atom within the molecule [32].
The protein stability was determined by Schrodinger BioLuminate (version 1.0, Schrodinger, LLC, New York, NY). Structure analysis was performed using Have (y)Our Protein Explained (HOPE) developed by the Centre for Molecular and Bio-molecular Informatics (CMBI), Department of Bioinformatics, Radboud University [33]. This program was used to observe the impact of mutant variants on the factor VIII protein domains. The analysis was based on the parameters for functional contacts like metal, DNA, hydrogen bonds, ionic interactions, and structural areas together with motifs and domains including transmembrane domains. The last three parameters are very important to acquire factual data of actual protein structure resulting in annotated information in UniProtKB utilized by prediction by DAS servers [34]. Additionally, we also compared the predictions of the functional effects that were determined by the SNAP [35], this process provides binary classifications (impact/neutral) accompanied by a more detailed score. SNAP scores differ from -100 (strongly predicted as neutral) to 100 (strongly predicted to alter function); the distance is directly related to the binary determination boundary (0), which measures the reliability of the impact [36]. The structure primarily bases the mutant stability predictions on a protein of unknown structure. In the mutated form, the place of the mutated residue is specified, in place of wild type amino acid. Amino acids residue scanning was performed to fix polar and neutral residues for analysis of the protein stability and solvent accessibility. Furthermore, SWISS-PROT and CCP4 (QTMG) analyzed the structure conformational changes of factor VIII protein domains.
| Results | ▴Top |
Analyses of mutations in factor VIII gene by capillary sequencing
The clinical symptoms of these patients with hemophilia A were shown in Table 1. The factor VIII samples which were tested negative for inversion 22 and inversion 1 were used for capillary sequencing. The mutations found in Saudi Arabian patients in factor VIII gene are listed in Table 2. Twenty-one DNA samples of Saudi Arabian patients with hemophilia A were sequenced for factor VIII gene. Out of 21 DNA samples we have screened, we have found three novel mutations, a novel mutation (c.99 G>T, p. (W33C)) was present in a 1-month-old male patient, with 1 to < 5% of factor VIII levels (moderete hemophilia A) with episodic bleeding. He was receiving plasma-derived factor VIII concentrate on demand. This patient did not have a history of joint disability or joint pains, and also did not have factor VIII inhibitors in his blood. This G>T mutation occurred in codon TGG of tryptophan (W) to change into TGT cystiene (C) and this amino acid is present in A1 domain of factor VIII. This results in the aromatic hydrophobic amino acid to change into hydrophobic aliphatic amino acid. In Figure 1, the electrophoregram shown in panel C is for the wild type control and in panel D, for the patient with mutation in exon 1. In panel B, PCR gel picture is shown for exon 1 product size of 638 bp, and the acutal exon 1 size is 344 bp, transcript ID: F8-001 ENST00000360256.8 (Ensembl.org).
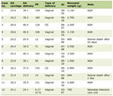 Click to view | Table 1. Representative Hemophilia Patient’s Clinical Characteristics |
Click to view | Table 2. Summary of Mutations Detected in Saudi Arabian Hemophilia Patients |
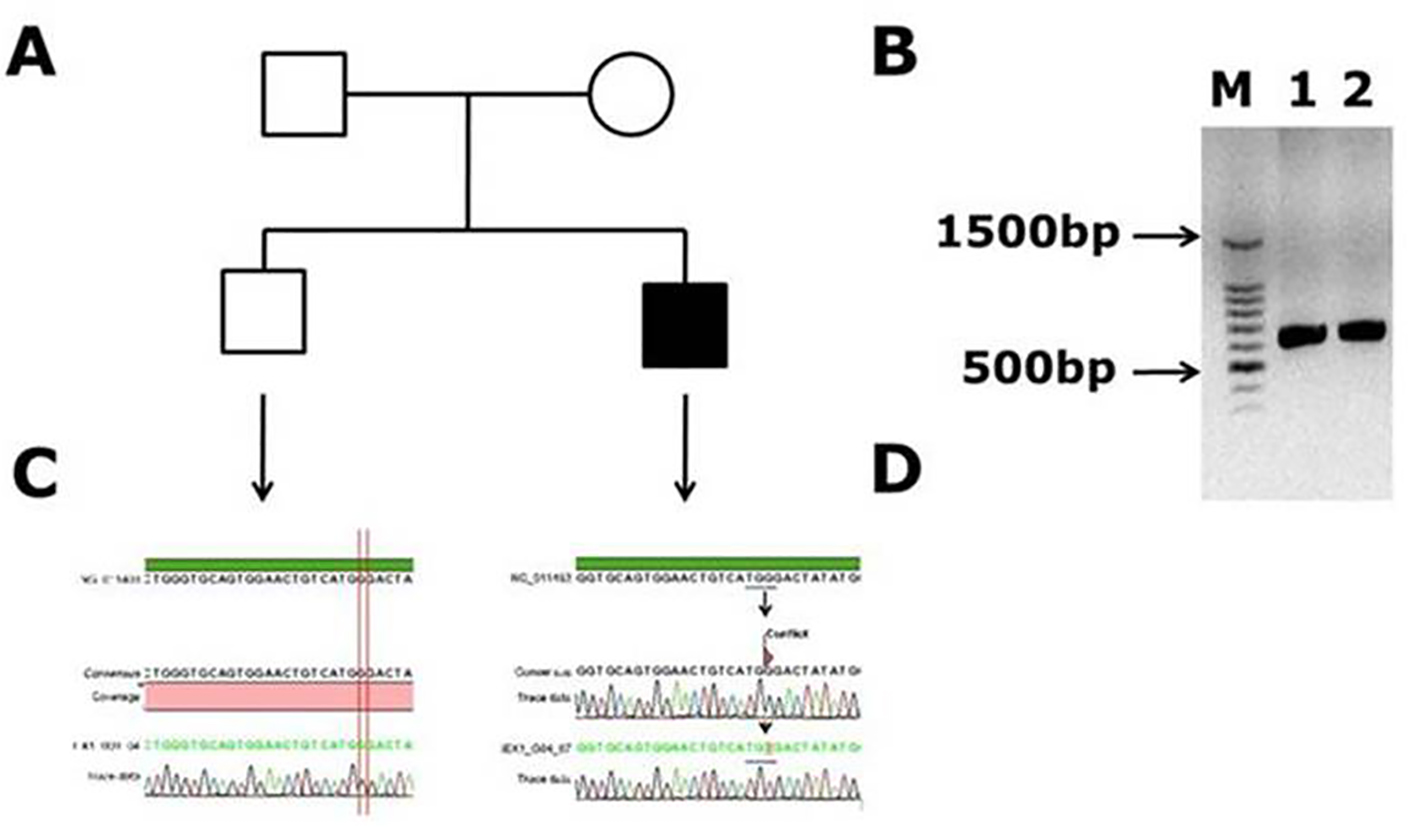 Click for large image | Figure 1. Novel missense mutation identified in exon 1 of the factor VIII gene. (A) Family pedigree. (B) PCR gel image of exon 1 in factor VIII showing single specific band of exon 1 product size 638 bp in patient and WT (lanes 1 and 2). (C) Representative electrophoregram showing WT genotype. (D) Representative electrophoregram showing the mutation (conflict) in exon 1. |
Another novel muttaion was identified in one male patient in exon 14, and it is a frameshift mutation c.2138 delA, p. (N713Tfs*9) causing blood coagulation factor VIII deficiency in a Saudi Arab family. This deletion of A in the codon AAC of aminoacid aspargine (N) changed to codon ACT and codes for threonine (T) and inturn a frameshift occurs in the coding of nine amino acids further and a stop codon (TGA) at ninth amino acid leads to transcriptional termination and truncated protein. The electrophoregram is shown in Figure 2. This patient was a 1-month-old male with episodic bleeding and mild hemophilia A symptoms with factor VIII levels of 5-40%, he did not have any symptoms of joint pains and factor VIII inhibitor also was not present. This pateint was undergoing treatment with plasma-derived clotting factor concentrate. In Figure 2, in panel B, PCR gel image of exon 14 of factor VIII shows single specific band of product size, 352 bp in patient and WT (lanes 1 and 2). As exon 14 is very large, multiple primers were designed for this exon; in panel C, the electrophoregram is shown for the wild type control, and in panel D, for the patient with mutation in exon 14. In one patient, a novel mutation at exon intron boundary of exon 23 and intron 22 was found. This patient was a 1-month-old male, with moderate hemophilia with baseline factor VIII levels of 1 to < 5%. This pateint had chronic joint pains and was undergoing treatment with plasma-derived clotting factor concentrate, and tested negative for factor VIII inhibitor. This mutation G>A is present at the splicing site of the exon 23 (Fig. 3).
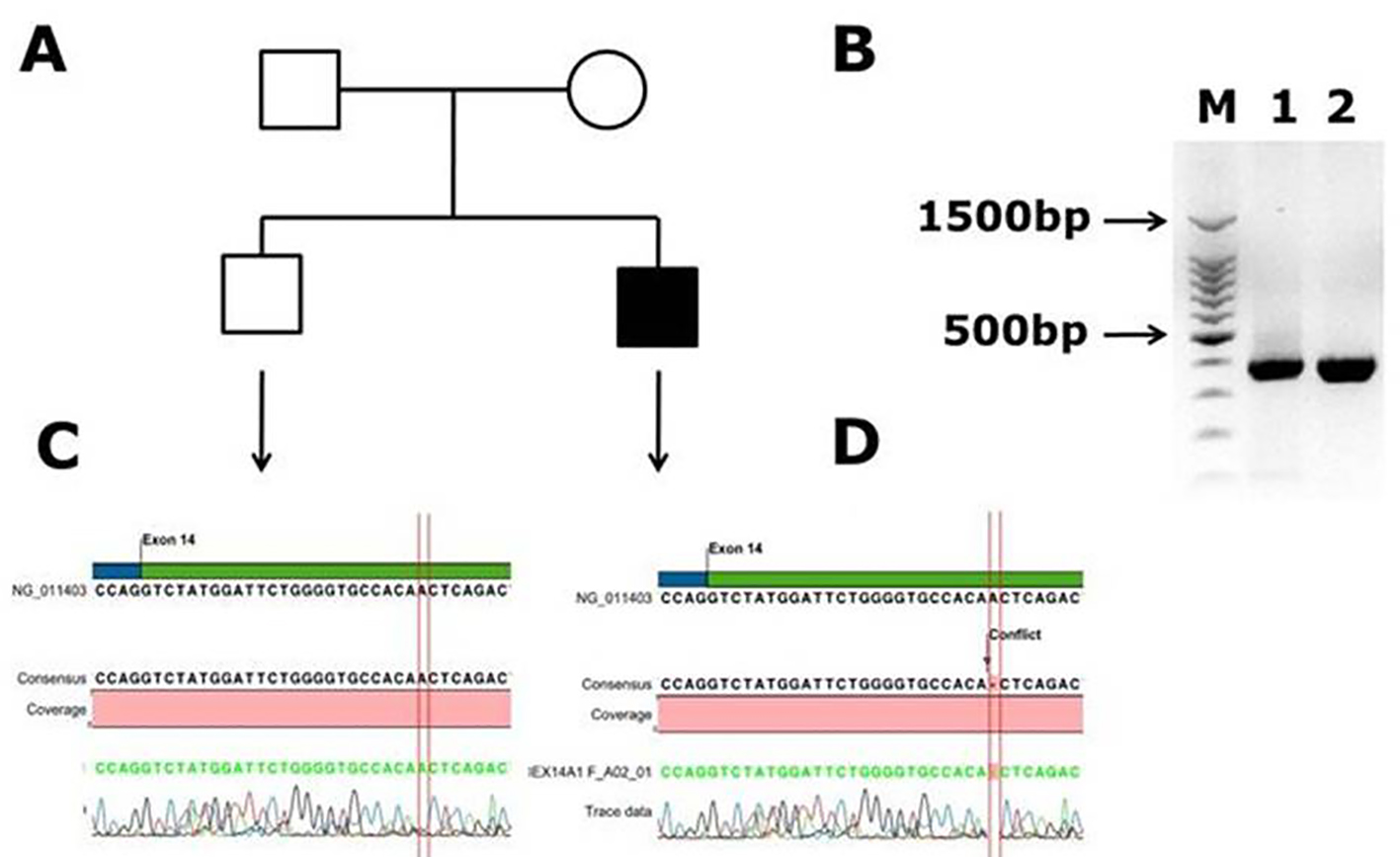 Click for large image | Figure 2. Identification of novel frame shift mutation in coagulation factor VIII c.2138 delA, p. (N713Tfs*9) causing hemophilia A in Saudi Arabian patient. (A) Family pedigree. (B) PCR gel image of exon 14 of factor VIII showing single specific band of product size 352 bp in patient and WT (lanes 1 and 2). As exon 14 is very large, we have designed primers of small fragments of exon 14. (C) Representative electrophoregram showing WT genotype. (D) Representative electrophoregram showing the mutation (conflict) in exon 14 of factor VIII gene. |
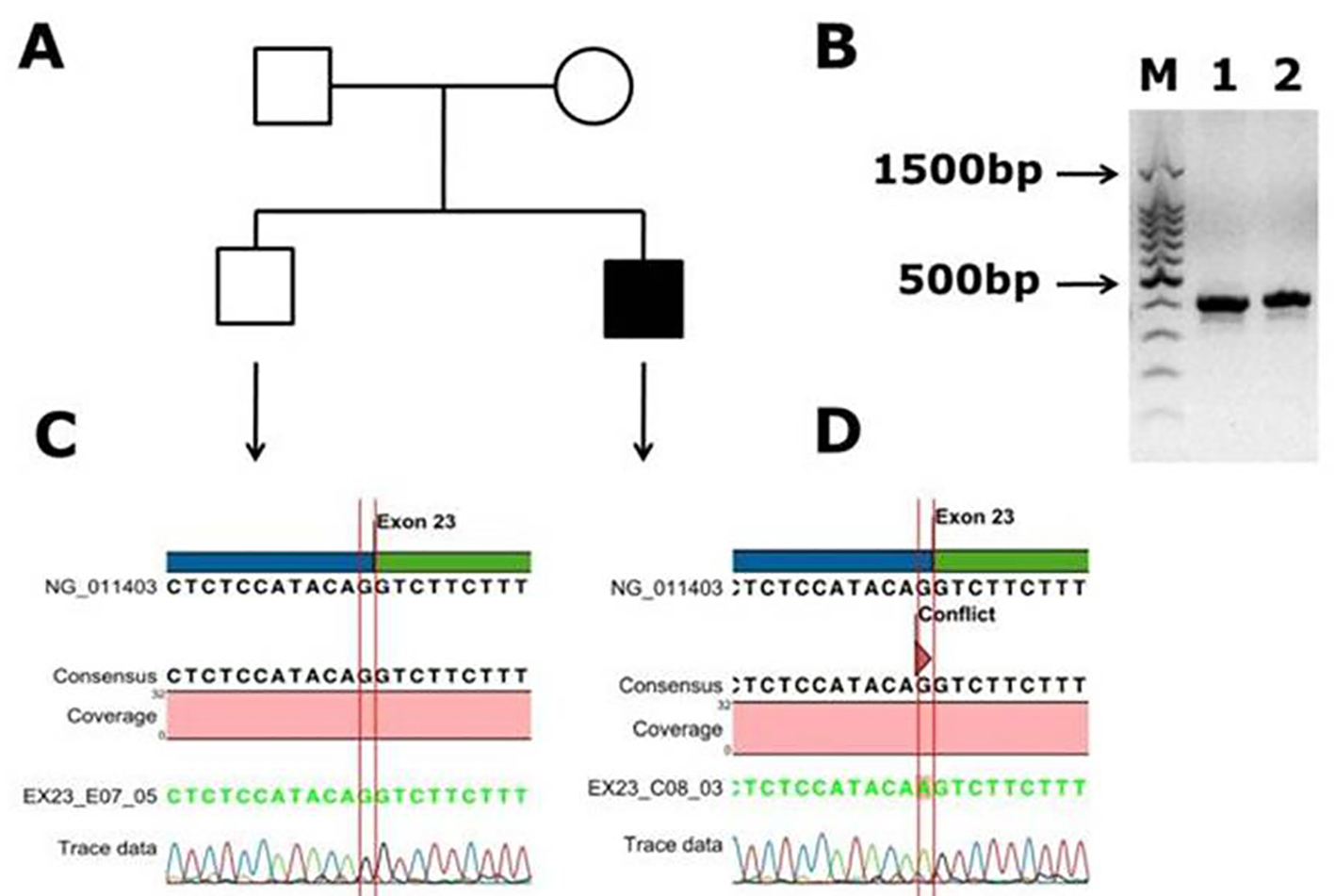 Click for large image | Figure 3. Identification of a novel splicing mutation c.6430 -1 G>A (splicing acceptor site) of factor VIII in a Saudi Arabian patient. (A) Family pedigree. (B) PCR gel image of exon 23 of factor VIII showing single specific band of product size 416 bp in patient and WT (lanes 1 and 2). (C) Representative electrophoregram showing WT genotype. (D) Representative electrophoregram showing the mutation (conflict) at exon intron junction in exon 23 of factor VIII gene. |
In one patient who was a 2-month-old male, we found a previously reported mutation (c.760 A>G, p. (N254D)) in exon 6 [21]. This patient had chronic joint disability (pain in right knee), he had factor VIII levels of less than 1% and under treatment with more than one product. In exon 12, a reported mutation c.1804C>T, p. (R602*) was identified in one patient. This patient was a 1-month-old male with episodic bleeding, he had factor VIII levels of less than 1% and was treated with more then one product for hemophilia A. Also another known mutation was found in factor VIII gene, in exon 12, (c.1836G>C, p. (R612P)) in two patients. One patient had pains in both knees and another patient had chronic joint disabilities, both patients had factor VIII baseline levels of < 1% (figures are not shown for these known mutations).
Analyses of mutations in factor IX gene by capillary sequencing
The clinical symptoms of these patients with hemophilia B are shown in Table 1. We have screened 42 DNA samples for all factor IX exons by capillary sequencing. The mutations found in Saudi Arabian patients in factor IX gene are shown in Table 2. Out of 42 DNA samples tested, one novel mutation was found in one patient, and five are reported mutations found in 11 individuals. The novel mutation found in this study was a missense mutation c.855G>C, p. ((E285D); GAG>GAC). The patient was a 1-month-old male with episodic bleeding and severe form of hemophilia B, he was unable to flex his knees, elbows and forearms, and he was undergoing treatment with factor IX concentrates. The electrophoregram is shown in Figure 4; in panel B, the PCR gel is shown for exon 8 of factor IX with specific band of product size, 707 bp. Panel C is electrophoregram showing WT genotype; panel D is electrophoregram showing the missense mutation (conflict) in exon 8 of factor IX.
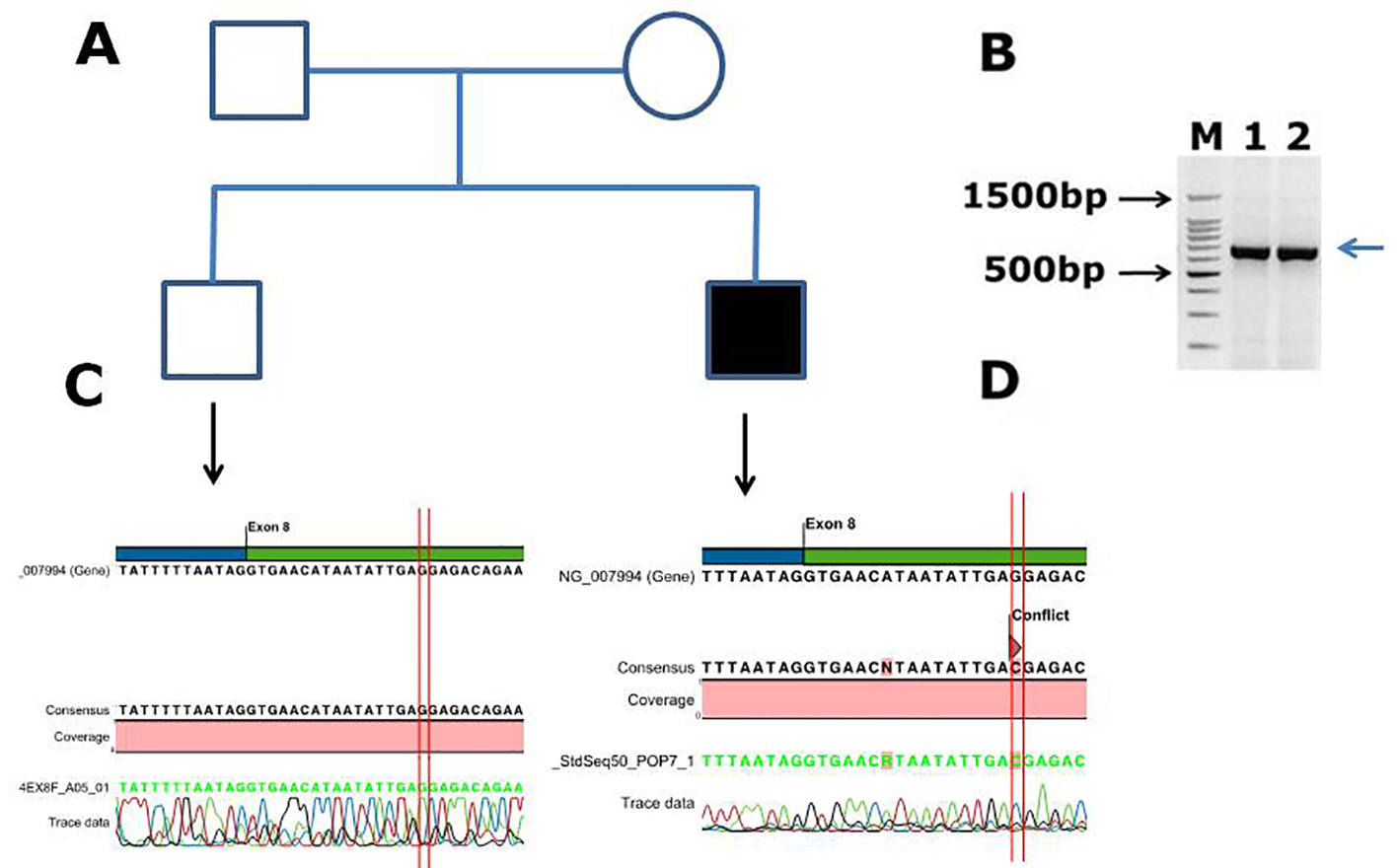 Click for large image | Figure 4. Identification of a novel missense mutation in coagulation factor IX c.855G>C, p. (E285D) causing hemophilia B in Saudi Arabian patient. (A) Family pedigree. (B) PCR gel image of factor IX exon 8, showing single specific band of product size 707 bp in two patients (lanes 1 and 2). (C) Representative electrophoregram showing WT genotype. (D) Representative electrophoregram showing the missense mutation (conflict) in exon 8 of factor IX. |
In exon 4 of factor IX, in the EGF-1 domain, a known point (missense) mutation c.316G>A, p. ((G106S); GGC>AGC) was found in one patient. In six patients out of the 42 DNA samples, we found missense mutation c.580G>A, p. (A194T), a polymorphism in exon 6 in the activated peptide of factor IX. In exon 8, a known mutation (CGA>TGA) in the serine protease domain of factor IX, c.880C>T, p. (R294*) was found in two patients, and both had chronic joint pains, and severe form of hemophilia B. Two patients have point mutations c.1136G>A, p. (R379Q; (missense)) and c.1025C>T, p. (T342M) (ACG>ATG; missense) in exon 8. These patients had severe form of hemophilia B. Summary of the mutations is shown in Table 2, but figures for the electrophoregrams and PCR gel pictures of these known mutations were not shown here.
Factor VIII MD simulation studies
Crystal structure modeling of factor VIII coagulation domains (PDB 2R7E) is shown in Figure 5. Positions of the mutation studied are indicated by arrows, and this position of the mutation in close-up view with the mutated and nearby residues is shown as ball and stick models as dashed circles respectively. The W 33 to C mutation located in the interface of the A1 domain. The annotation for the missense mutation c.99 G>T, p. (W 33 C) within the gene on X chromosome was performed. Protein structure of the factor VIII domain (PDB 2R7E) coding regions was retrieved from the protein data bank (PDB) database, and we resolved the structure from UniProtKB (P00451 (FA8_HUMAN)) (Fig. 5A, B). This structure was typically from five domains (A1-A2-A3; C1-C2), and two chains A and B. But our targeted sequence in which mutation was found aligned with only the “chain A” in the domain A1. The mutation W 33 to C was within loop of the factor VIII associated protein domain “chain A” structure. The corresponding positions were viewed individually near mutation site to examine the altered model structures utilizing CCP4 (QtMG). The predicted protein evaluation was determined using a Ramachandran plot with a favored region (98.0%), and allowed area about 2.0%, and an outlier region of 10.4%. We found greater than 90% of the residues in the most favored regions, which showed that the quality of the structure was comparable to the template [37].
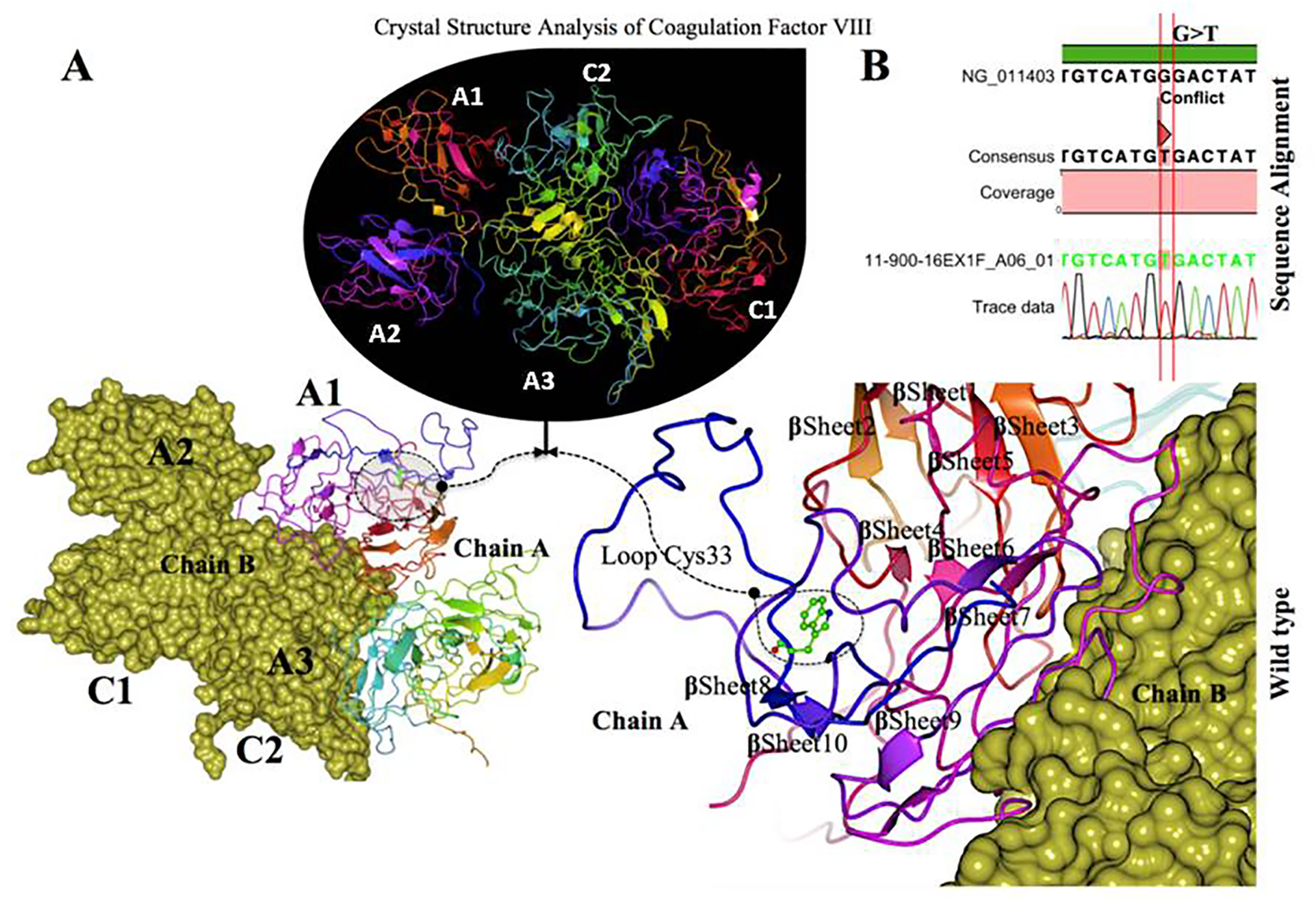 Click for large image | Figure 5. Crystal structure of coagulation domains of hemophilia factor VIII protein and MD simulation. (A) The crystal structure of the domains of factor VIII (PDB ID 2R7E), the individual domains are labeled and also in different colors (A1) in magenta, (A2) shown in blue, (A3) shown in yellowish green, (C1) shown in pink, and (C2) shown in bluish green. The molecular surface of factor VIII domains (A2) and (A3) are in chain B (yellow). Overview views positions of mutation with nearby residues shown as ball and stick models in dashed circle. The (Try33Cys) mutation located in the interface of (A1) domain, a tertiary structure of the wild type chain A of factor VIII protein showing the mutation (W 33 C) green on a loop region near with alpha helix 10. (B) The electrophoregram showing the sequence alignment the patient sample with the wild type for this mutation G>T. |
Factor VIII protein MD simulation is shown with truncated boundary explicit water solvated and hydrogen atoms in Figure 6. Tertiary structure of the factor VIII protein shows hemophilia causing mutation, i.e. gray cysteine 33 (wild type, green tryptophan 33), on loop region near with alpha helix10 modeling (Fig. 6A, B). The visual inspection also identified the side chain of a histidine residue involved in the hydrogen bonding with surrounding molecules (Fig. 6C). The residues were scanned, and loop mutation analysis was performed for factor VIII gene. W 33 in factor VIII and another thermo stable site (cysteine 33) were targeted as critical residues and were substituted using Schrodinger (BioLuminate). The energy minimization of factor VIII before the mutation was performed at -421,971.904 kJ/mol, which decreased to -397,569.203kJ/mol after mutation, as calculated by using BioLuminate program. The energy minimization was based on the set of selected polar and non-neutral residues for these screenings. We fixed the refinement cut-off of 0.00 Å with an implicit solvent minimization. The result of decreasing the minimized energy value of the loop: W 33 C showed that the substitution of W 33 to C may contribute to improved compactness and may increase protein folding. Further loop mutation was based on the implicit solvent according to MD simulations (Fig. 6A, B, C). During these simulations, proteins continuously fold and unfold and provide considerable insight into this process [38]. Compared to the results at a temperature of 300 K, the deviation of the factor VIII protein increased from 2.0 Å in 10 ns, in contrast, the deviation of mutated protein was maintained at 1.3 Å until the end of the simulation (t = 10 ns), which showed that it had reached its folded state. The small peak at 1.8 ns indicated that W 33 C stabilized the structure. Our results showed that the W 33 C structure was stable and could maintain its conformation at 300 K, at a pressure bar of 1.01225 with a surface tension 4,000.0 Å, in a total simulation time (ns) of 1.2/elapsed 0.0, and recording interval (ps) energy of 1.2. However, there were subtle changes that were observed between wild type and mutant factor VIII, as revealed by the superimposed structures with an RMSD value of Å, which were determined using MOE. Tryptophan 33 of the factor VIII protein showed a higher solvent accessible surface area than the mutant cysteine 33 inside the protein structure. The structural replacement of tryptophan 33 with a less hydrophobic residue cysteine 33 residue inside of the protein structure was able to increase the compactness. In a further analysis of annotated predicted solvent accessibility and pre-calculated packing density, W 33 C was found to increase the density and lessen the internal cavities compared to the wild type form. Thus, substitution of a residue (W 33) with C 33 enhanced the packing and compactness of the mutant structure and decreased the internal residue in the protein structure.
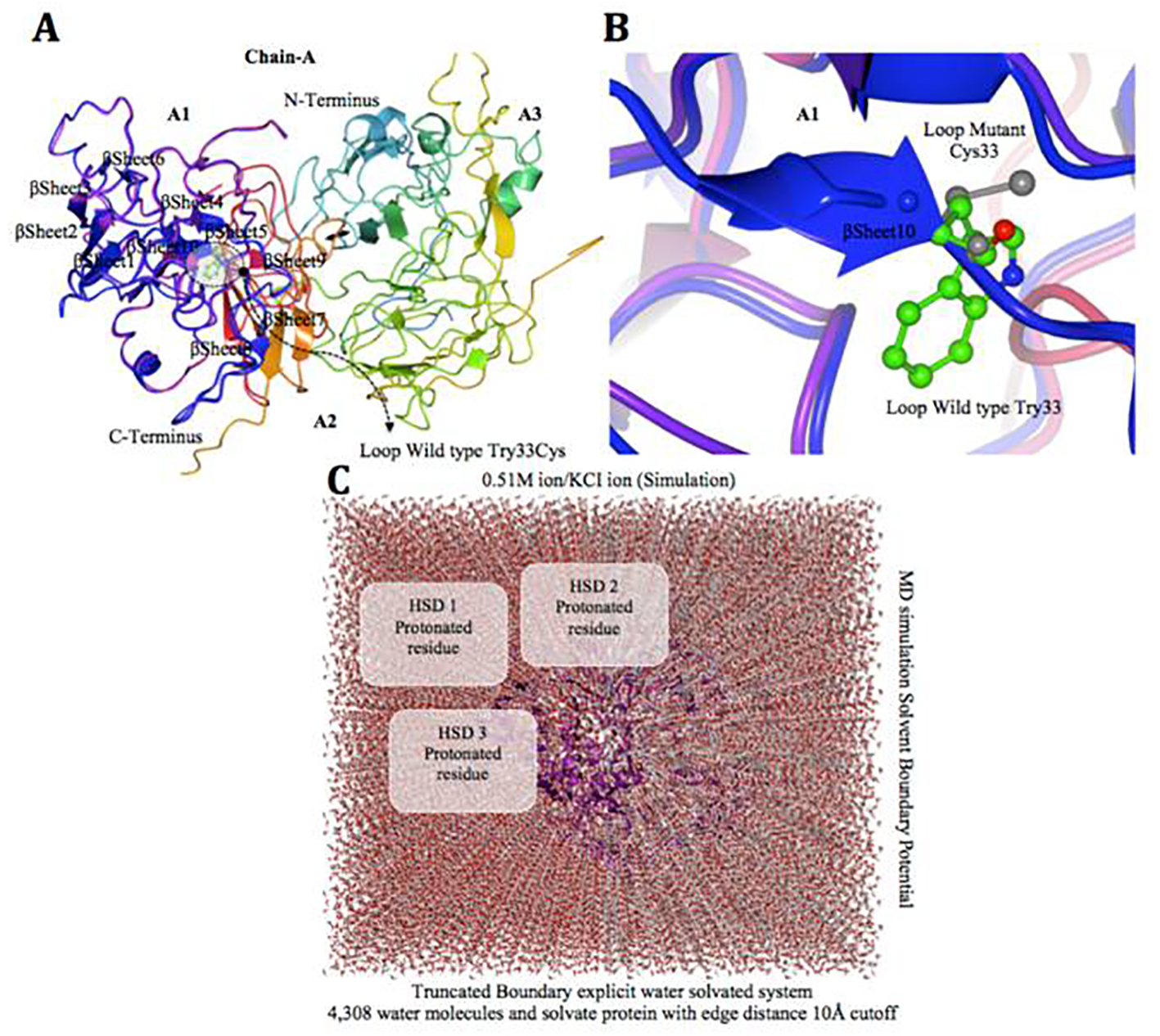 Click for large image | Figure 6. Factor VIII protein MD simulation showing truncated boundary explicit water solvated and hydrogen atoms. (A and B) Tertiary structure of the factor VIII protein showing amino acid mutation, i.e. gray cysteine (C 33), wild type green tryptophan (W 33), on loop region near with alpha helix 10 modeling. (C) The visual inspection also allow to identify the side chain of a histidine residue involved in the hydrogen bonding with surrounding molecules and the δ nitrogen of the histidine (HSD) is protonated. The MD simulation system used in calculations is: water box surrounding the entire protein (middle). |
Factor VIII protein conformational changes of RMSD and root mean square fluctuation (RMSF)
Trajectories over 10 ns were analyzed by calculating the RMSD of factor VIII at 300 K. At the beginning of the trajectory (t = 0), RMSD of factor VIII value of 0.6 Å indicated the movements occurring during the thermalization and equilibration periods. At 300 K, the deviation value increased rapidly during the first 3 ns and maintained the deviation until the end of simulation (t = 10 ns). Meanwhile, change in tryptophan to cysteine showed rapid increase of the deviation value (1.6 Å) until 8 ns and decreased to the end of simulation, and this protein structure was not stable and did not reach the folded state until end of the simulation at 50 °C. From these result, it showed that W 33 C structure was stable and could maintain the conformation at 300 K (60 °C), meanwhile factor VIII could be stable at 300 K/50 °C. It is proven that the substitution of W 33 with C had increased the thermal stability of protein structure in mutant regarding the longer stay in the folded state at higher temperature. Furthermore, RMSF of residue factor VIII (W 33 C) was used to calculate over the trajectory for overall flexibility of the system at temperatures (300 K). A slight fluctuation was observed in factor VIII protein sequence at targeted mutated region, which was at the flexible region of factor VIII. The flexible region consists of loop mutation that was located in near the α-sheet 10 of factor VIII domain A1. The fluctuation of flexible region increased the flexibility at higher temperature (300 K/60 °C). The critical point mutation increased the protein folding and compactness due to less flexibility of flexible region. Trajectories of MD simulations over 10 ns of W 33 C structure were sampled at 50 and 60 °C to analyze the differences of structural stabilities. The N-terminal of factor VIII protein was shown in the structures before and after the simulations (10 ns) as expressed by different temperatures. The large fluctuation of flexible region would trigger an unfolding process and consequently denature the protein [39].
Effects of single amino acid substitutions and protein structural loop stability due to W 33 C mutation
The effects of critical point missense mutation towards thermodynamic stability were used to study by Bioluminate and MOE. The factor VIII W 33 C was analyzed by MD simulation at various temperatures starting from 300.0 K at time setup 1.0 fs. The pressure bar was increased by the coupling factor temperature at 100.0 fs. It meanwhile increased the stability in loop from 300 K. The thermal transition curve obtained by temperature dependence at 1.0 fs also supported that mutant increased the thermostability. The temperature is directly related to protein stability. As such, it was assumed that the substitution of less hydrophobic residue (W 33) to more hydrophobic residue (C) increased the internal hydrophobicity of W 33 C to maintain the structural stability at a high temperature. Altering amino acid 33 from W to C yields excessive-folding free energy (ΔΔG = -3.704 kcal/mol), that includes a destabilizing impact on factor VIII domain A1 structure while compared in both wild and mutant. Moreover, the mutant tertiary structure of factor VIII domain which alters from W to C at amino acid position 33 causes significant perturbations to folding particularly region with correlation coefficient of 0.8 between predicted and measured stability changes; in cross-validation, after exclusion of 10% outliers, the best value was 0.67 (Fig. 7A, B).
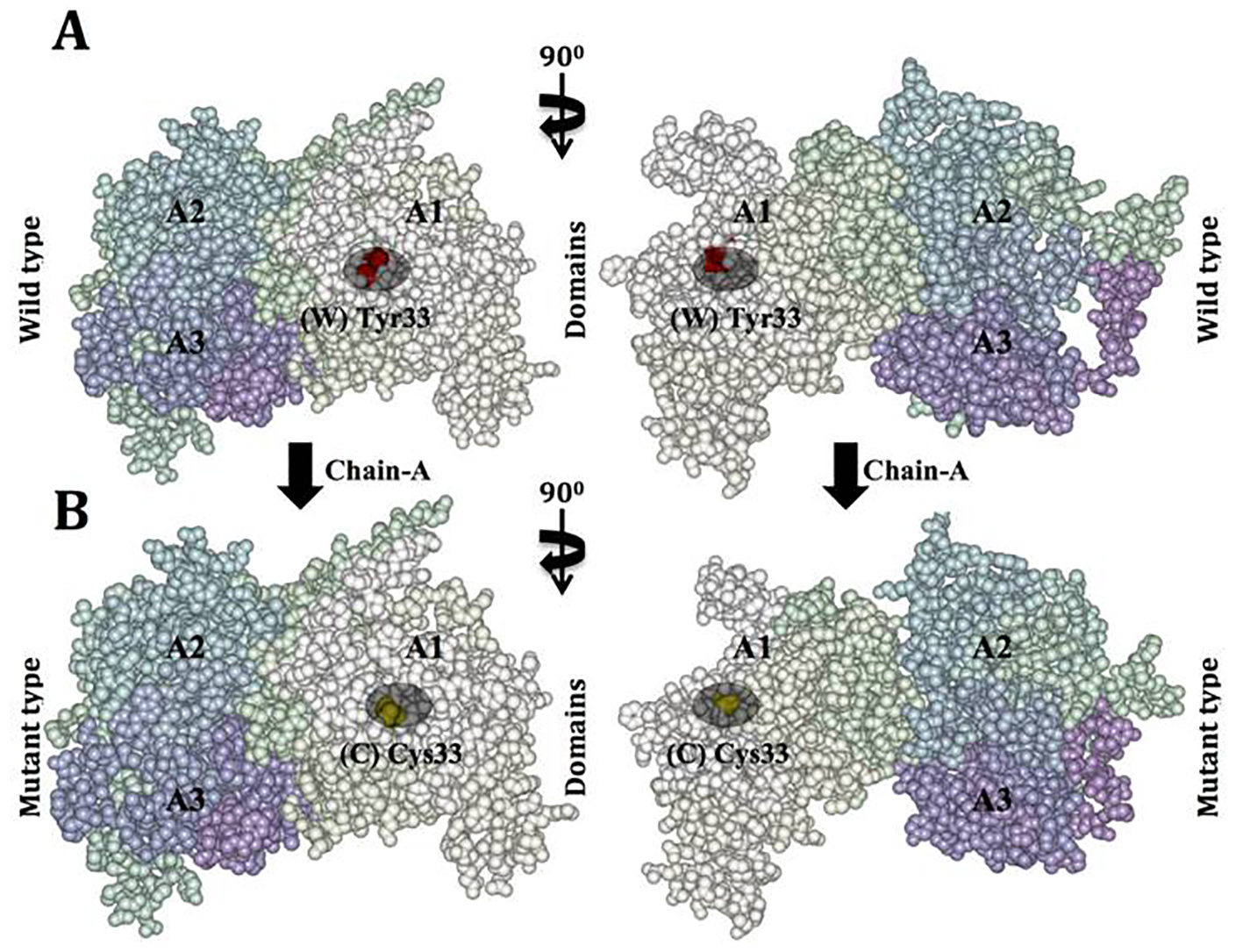 Click for large image | Figure 7. Van der Waals surface spacefill model of factor VIII protein A1, A2, and A3 domains showing solvent-exposed mutation on the surface. In this surface representation of quaternary structure of human factor VIII domains (A1, A2 and A3), the region of hemophilic mutation site is depicted as a cluster. (A) Chain A contains domain A1-A2-A3 labeled, in domain A1 the native residue W 33 is represented as (W) in red cluster. (B) Chain B surface model contains domain A1-A2-A3 labeled. Domain A1 has a mutated residue C 33 is represented in yellow color cluster (C). |
In Figure 8, MD simulation of factor VIII, c.2138delA, p. (N713Tfs*9) novel frame shift mutation is shown. Overview of the protein in the form of ribbon was presented in panels A and B. The protein is colored by element α-helix (blue), β-strand (red), turn (green), 3/10 helix (yellow) and random coil (cyan), and the other molecules in the complex are colored gray. In the panel B, overview of the protein in ribbon form clearly shows the side chain of the mutated residue in magenta shown as balls socket model, and the protein is colored gray; in panel C, close-up of the mutation c.2138delA, the protein was shown in gray color, the side chains of both the wild type and the mutant residue are shown and colored green and red respectively indicated with arrow marks. In panel D, close-up of the mutation (seen from a slightly different angle), the protein is colored gray; the side chains of both the wild type and the mutant residue are shown and colored green and red, respectively.
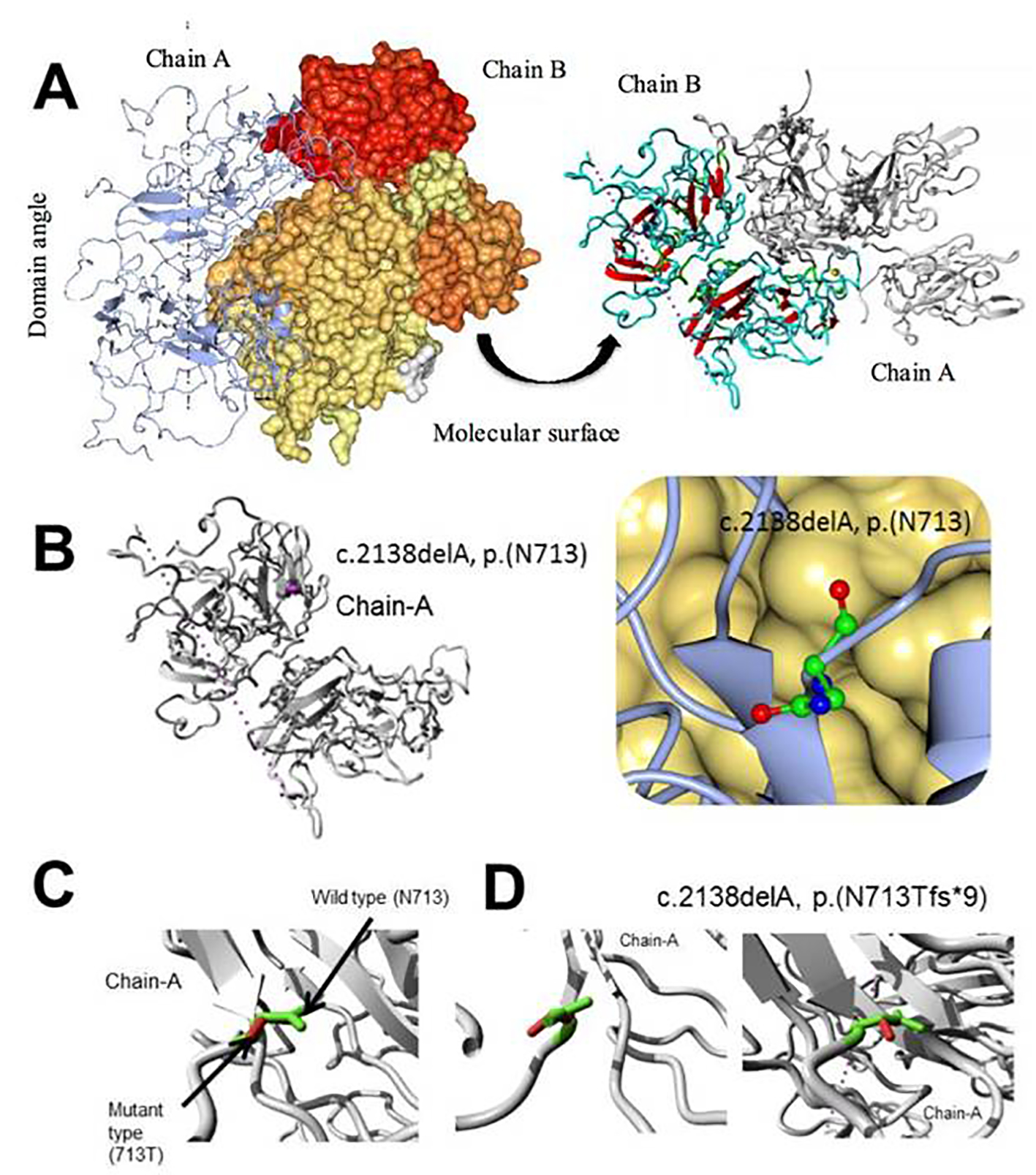 Click for large image | Figure 8. Molecular dynamics simulation of factor VIII, c.2138delA, p. (N713Tfs*9) novel frame shift mutation. (A) Overview of the protein in ribbon-presentation. The protein is colored by element α-helix (blue), β-strand (red), turn (green), 3/10 helix (yellow) and random coil (cyan). (Other molecules in the complex are colored gray). (B) Overview of the protein in ribbon-presentation. The protein is colored gray; the side chain of the mutated residue is colored magenta and shown as small balls. (C) Close-up of the mutation. The protein is colored gray; the side chains of both the wild type and the mutant residue are shown and colored green and red, respectively. (D) Close-up of the mutation (seen from a slightly different angle). The protein is colored gray; the side chains of both the wild type and the mutant residue are shown and colored green and red, respectively. |
| Discussion | ▴Top |
We report here novel factor VIII and factor IX mutations in Saudi Arabian hemophilia patients, and molecular simulation results of the mutated domains of factor VIII protein using computer modeling. The factor VIII novel mutations identified were c.99G>T, p. (W33C), c.2138 DelA, p. (N713Tfs*9) and a novel mutation at splicing acceptor site in exon 23 c.6430 - 1G>A. In factor IX, we found a novel mutation c.855G>C, p. (E285D). These novel mutations were not reported previously in databases such as factorviii-db.org or factorix.org. To our knowledge, this is the first study reporting this novel mutation c.99G>T, p. ((W33C; TGG>TGT)) in exon 1 of factor VIII gene. However, a previous study by Tavassoli et al [40] has reported a different mutation in this amino acid coding tryptophan 33 to glycine 33 (c.97 TGG>GGG, p.(W 33 G)). This W33G mutation was also associated with moderate form of hemophilia A in that German patient [40]. The novel mutation (c.2138 DelA) we found in this study causes a frameshift in exon 14. This frame shift mutation detected in the exon 14 of the factor VIII resulted in a stop codon and is expected to result in a truncated protein. Also, a mutaion in this codon (c.2138 A>T) was reported previously causing N713 to change into isoleucine and this muttaion also was associated with mild type of hemophilia [41]. Exon 14 of factor VIII gene is larger than 3,000 bp, and many deletions, insertions, and point mutations were reported in this exon in European, Jordanian Tunisian and Lebanese population [42-45].
The mutation identified (c.760 A>G,p. (N254D)) in exon 6 in this study was previously reported. This A>G mutation in codon 760 changes AAT to GAT, and in turn neutral amino acid aspargine becomes negatively charged aspartic acid. This mutation was reported previously in five patients [21, 46] and was associated with severe form of hemophilia A, and our results also confirmed this. In exon 12, a reported mutation c.1804 C>T, p. (R602*) was identified, and this mutation in arginine codon (CGA>TGA) makes a stop codon causing a truncated protein production. This mutation is reported to be associated with severe to moderate form of hemophilia A, and our results also confirmed this. Also another known mutation was found in this exon (c.1835G>C, p. (R612P)) in two patients. This mutation in the A2 domain of factor VIII causes positively charged hydrophilic arginine to code for the hydrophobic neutral amino acid proline. This mutation (CGC>CCC) is associated with severe form of hemophilia A [21]. One of these male patients had factor VIII inhibitor, and had chronic joint disabilities; both patients had severe form of hemophilia A. The novel mutation at splicing acceptor site in exon 23 c.6430 - 1G>A was present 1 base before the exon 23 start (before the first codon of exon 23) amino acid valine (GTC) sequence. The functional effect of this mutation by SIFT and PolyPhen-2 is shown as “tolerated”; however, as this patient was not available for fresh blood collection for RNA isolation, we could not check whether this mutation is affecting the transcriptional process or not.
More than 1,000 mutations have been reported for factor IX to date (Factorix.org; http://hadb.org.uk). Literature review shows that there are no studies reporting for factor IX mutations in Saudi population, or in the Middle Eastern population except from Iran, by Karimipoor et al, and Enayat et al [47, 48]. Our study is the first study of molecular genetic analysis of the factor IX gene in Saudi Arabian patients. In exon 4 of factor IX, in the EGF-1 domain a point (missense) mutation was reported previously in 74 patients from countries like India, China, Canada, USA, and South American [49]. This mutation c.316G>A, p. (G106S), was also found in Saudi Arabian patient. This 1-year-old patient had a mild form of hemophilia, and six individual patients were found to have missense mutation in c.580G>A, p. (A194T) in exon 6 in the activated peptide of factor IX gene. Only one of these six patients had factor IX inhibitor present. This polymorphism was previously reported in two patients from India so far [50]. This mutation is associated with severe form of hemophilia.
Factor IX exon 8 is very large (1.9 kb), and it codes for 181 amino acids. Several mutations were reported in this exon previously. We have identified one novel mutation in c.855G>C, p. (E285D) in exon 8 of the factor IX gene, and this is a missense mutation in the serine protease domain. This mutation causes the change in the amino acid glutamate to aspartic acid. Glutamic acid is a hydrophobic amino acid, whereas aspartic acid is a hydrophilic amino acid. Glu285 is an exposed residue; the surface accessibility from the modeled factor IX structure is high for this, whereas, aspartate 285 is a buried residue, and the surface accessibility for this amino acid is low. A known mutation in this serine protease domain of factor IX, c.880C>T, p. R294* (ATG stop codon), was reported previously in 70 patients [51, 52]. This frame shift mutation detected in exon 8 of factor IX resulted in a stop codon and is expected to result in a truncated protein. It was reported to be associated with moderate to severe form of hemophilia. One known mutation c.1136G>A, p. (R379Q) was identified in exon 8 of the factor IX gene. This point mutation (missense) in the serine protease domain causes positively charged amino acid (arginine) to change to a neutral amino acid (glutamine). This mutation was also reported in Chinese, Canadian, Americans, Turkish, European, and Indian, it is associated with moderate to severe hemophilia, and it is reported in 69 patients [50, 53]. Also another known mutation was identified in exon 8, and this is a point mutation c.1025C>T, p. (T342M). This mutation caused a change in hydrophilic to hydrophobic amino acid. This mutation was reported previously in 128 patients from China, Canada, USA, Turkey, South American, and India. It is associated with mild to severe form of hemophilia [18, 54].
The novel mutations identified in this study in factor VIII and factor IX genes had not been previously assessed computationally for structural and functional studies by MD simulation. This is the first study that reports the predictions of functional and structural effect of the annotated mutations in factor VIII protein in exon 1 and in exon 14, using high throughput evaluation of MD simulation on prediction of factor VIII protein domain before and after mutation. The protein structural analysis was performed considering this particular mutation, located at factor VIII gene and compared with the predicted native protein. In addition, for the native and mutant protein models, the amino acid residue and its secondary structure were determined based on the solvent accessibility for the confirmation of the protein stability at point mutation. A single mutation that causes an amino acid alteration can drastically modify the stability of the resulting protein structure; therefore, the modeling of protein structural data will likely be a requisite for the understanding of protein functionality [55]. Protein function will typically be associated with evolutionarily conserved residues [56]. Modifications in the folding free energy upon mutation (ΔΔG) support the notion that a mutation causes disease primarily because it damages an important protein function. In this report, crystal structure of the factor VIII protein domain PDB2R7E with this novel mutation in exon 1 (W 33 C) has been determined, showing significance of this mutation in A1 domain. The most crucial structural element in this loop appears to be the specific positioning of residue cysteine 33 (C 33), which is in close coordination with the helix 10. Additionally, we have examined the native and mutant protein structure for solvent accessibility and secondary structural changes. We have found that this novel mutation is likely to impact the protein structure. We have identified the potential mutation and proposed structural model for the mutant protein domain A1, and compared that with the wild type domain. We also analyzed native and mutant domains for stability analysis, solvent accessibility and secondary structural analysis.
The mutation of tryptophan into cysteine at position 33 affects the factor VIII protein because of their differences in size, and hydrophobicity value. The original wild type residue and newly introduced mutant residue differ in these properties. In this case, the mutant residue was smaller than the wild type residue, and the wild type residue was more hydrophobic than the mutant residue. Moreover, the mutant tertiary structure of factor VIII domain which was altered by this mutation tryptophan to cysteine at amino acid position 33 causes significant perturbations to folding particularly region with correlation coefficient of 0.8 between predicted and measured stability changes; in cross-validation, after exclusion of 10% outliers, the best value was 0.67. Considerably bigger than average structural weaknesses were detected, that may contribute to the protein function and stability.
The wild type residue tryptophan was neutral and non-polar hydrophobic amino acid, and the mutant residue cysteine was uncharged (neutral) polar amino acid. The mutation changes the polarity in the amino acid residue; this can cause changes in solubility properties of the protein. Because the hydrophobicity of the wild type and mutant residue differs, this caused loss of hydrophobic interactions with other molecules on the surface of the protein. However, cysteine can form intra-chain or inter-chain disulfide bonds often occurring in proteins in its oxidized form known as cystine, and this modification will have an important role in altering the folded conformation of the protein. In its reduced (native) form this cysteine amino acid could play an important role with the conformational stability of factor VIII protein by either forming a non-native disulfide bond or simply by aiding covalent aggregation of the unfolded or misfolded factor VIII protein; however, this has to be established with wet lab analysis to understand the molecular mechanism behind the observed phenotype.
In this study, we explored the relationship between prediction consequences of novel mutation as determined by computational approaches based on recent findings from protein structural modeling and real phenotypes. The recent progress made in experimental 3D structure determination of domain organization of coagulation factor VIII by X-ray crystallography and modeling studies have made it possible to predict the effects of nsSNPs at structural level by mapping them on corresponding structures [57, 58]. There have been quite a lot of studies existing in literature validating the importance of single amino acid substitutions in hemophilia A and hemophilia B at activation cleavage sites, affecting factor VIII binding to von Will brand factor, factor VIII secretion and factor IX binding to factor VIII, respectively [59, 60]. In our study, the mutated residue cysteine 33 of factor VIII was located within a domain, annotated in UniProtKB as: “F5/8 type A 1; Plastocyanin-like 1”. It belongs to the multicopper oxidase family, containing 3 F5/8 type A domains, 2 F5/8 type C domains, and 6 plastocyanin-like domains [61]. Plastocyanin-like type 1, which can disturb this domain and abolish its function, a mutation to cysteine was found at this position. This variant differs from our mutation and can still be interesting and the reference of ExPASy site about this variant (VAR_028452). The mutation was located in a region with known splice variants described as: “In isoform 2” [62, 63]. Based on conservation scores, this mutation was probably damaging to the protein. Our MD simulation suggests the mutated residue was located on the surface of a domain with unknown function. This cysteine residue was not found to be in contact with other domains of which the function was known, nevertheless, contact with other molecules or a domain was still possible and might be affected by this mutation. This can cause repulsion between the mutant and neighboring residues. The mutation causes loss of hydrophobic interactions with other molecules on the surface of the protein.
Recently few reports from Arab and North African countries are appearing in literature for factor VIII and factor IX mutations. There are no conclusive hotspots mutations in these genes, except the inversion 1 and inversion 22 mutations in factor VIII. The accurate number of hemophilias A and B cases in Saudi Arabia so far is not clear [64], and very few reports of molecular analysis of factor VIII gene [20-22] and no factor IX mutation information was reported from Saudi Arabian patients. Our study is the first to report the mutations in factor IX gene in Saudi Arabian patients. In conclusion, we report here three novel mutations in factor VIII and one novel mutation in factor IX in Saudi Arabian patients. Our study includes a small population; therefore, a large study with more Arab countries is needed in order to determine the prevalence of various mutations in the Middle East. Identification of the causative mutations will increase our knowledge about the molecular mechanisms of hemophilia in this region. Most of the mutations detected in our study were deleterious, and these mutations correlated well with the severity of the phenotypes in the study. Presence of factor VIII and factor IX inhibitors was observed only in two patients with mutations, because all these patients were treated with plasma-derived clotting factor substitute. It has been reported that this therapy was associated with a low rate of inhibitor development [65]. Our results showed that two of the factor VIII novel mutations and the factor IX novel mutation were pathological and was clinically significant as identified by computational predictions SIFT and PolyPhen2. We have identified the important feature of the mutated domains of factor VIII through in-silico studies which support with clinical data. Our results suggest that these mutations have significant impact on the structure and function of the mutated factor VIII protein. We also observed whether the mutation is affecting the protein structure and function, using MD simulations by immersing them in a solvent using superior simulation parameters as well as energy minimization to analyze the simulation in terms of potential energies, structural fluctuations, coordinate stability and geometrical features.
Acknowledgments
We are indebted to the patients and their family members who participated in this study. The authors would like to thank the staff of Science and Technology Unit (STU) and Deanship of Scientific Research (DSR) at Umm Al-Qura University for the continuous support.
Conflicts of Interest
All authors agreed with the contents of the manuscript and all authors declare no conflicts of interest in publishing this manuscript.
Author Contributions
FAA conceived the idea, designed the research and analyzed data. MMT, MA, AB, and ZA performed experiments and analyzed data. FAA, MMT, NMB and ZA wrote the paper. HA and TO contributed vital reagents and clinical samples.
Grant Support
This work was supported by the National Science, Technology, and Innovative Plan (MAARIFAH) of the Kingdom of Saudi Arabia to Dr. Faisal A. Al-Allaf (grant code: 09-BIO920-10).
| References | ▴Top |
- Bowen DJ. Haemophilia A and haemophilia B: molecular insights. Mol Pathol. 2002;55(2):127-144.
doi pubmed - Forbes CD. The early history of haemophilia. In: Forbes CD, Aledort L, Madhok R. Eds. Haemophilia. London: Chapman & Hall, 1997; p, 3-6
- Mannucci PM, Tuddenham EG. The hemophilias - from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773-1779.
doi pubmed - Hoyer LW. Hemophilia A. N Engl J Med. 1994;330(1):38-47.
doi pubmed - Lillicrap D. The molecular basis of haemophilia B. Haemophilia. 1998;4(4):350-357.
doi pubmed - Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983-3996.
pubmed - Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801-1809.
doi - Graw J, Brackmann HH, Oldenburg J, Schneppenheim R, Spannagl M, Schwaab R. Haemophilia A: from mutation analysis to new therapies. Nat Rev Genet. 2005;6(6):488-501.
doi pubmed - Nielsen LR, Schwartz M, Scheibel E. Screening for mutations in the gene encoding factor IX. J Inherit Metab Dis. 1992;15(3):339-341.
doi pubmed - Anson DS, Choo KH, Rees DJ, Giannelli F, Gould K, Huddleston JA, Brownlee GG. The gene structure of human anti-haemophilic factor IX. EMBO J. 1984;3(5):1053-1060.
pubmed - Vehar GA, Keyt B, Eaton D, Rodriguez H, O'Brien DP, Rotblat F, Oppermann H, et al. Structure of human factor VIII. Nature. 1984;312(5992):337-342.
doi pubmed - Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry. 1985;24(14):3736-3750.
doi pubmed - Agus N, Yilmaz N, Colak A, Liv F. Levels of factor VIII and factor IX in fresh-frozen plasma produced from whole blood stored at 4 degrees C overnight in Turkey. Blood Transfus. 2012;10(2):191-193.
pubmed - White GC, 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85(3):560.
pubmed - Mannucci PM. Hemophilia and related bleeding disorders: a story of dismay and success. Hematology Am Soc Hematol Educ Program. 2002:1-9.
doi - http://www.moh.gov.sa/en/HealthAwareness/EducationalContent/Disease.
- Tarawah M, Owaidah T, Saleh M, Al-Kasim F, Al-Khaial T, Al-Zahrani H, Al-Zahrani A, Zolaly M, Al-Shahrani M, AlJassir F, Almomen A. Diagnosis and Management Guidelines of Haemophilia in Saudi Arabia. Journal of Applied Hematology. 2011;171-182.
- Saad S, Rowley G, Tagliavacca L, Green PM, Giannelli F. First report on UK database of haemophilia B mutations and pedigrees. UK Haemophilia Centres. Thromb Haemost. 1994;71(5):563-570.
pubmed - http://www.hgmd.cf.ac.uk/.
- Al-Allaf FA, Taher MM, Abduljaleel Z, Athar M, Ba-hammam FA, et al. Mutation Screening of the Factor VIII Gene in Haemophilia A in Saudi Arabia: Two Novel Mutations and Genotype-Phenotype Correlation. J Mol Genet Med. 2016;10:211.
doi - Owaidah TM, Alkhail HA, Zahrani HA, Musa AA, Saleh MA, Riash MA, Alodaib A, et al. Molecular genotyping of hemophilia A in Saudi Arabia: report of 2 novel mutations. Blood Coagul Fibrinolysis. 2009;20(6):415-418.
doi pubmed - Abu-Amero KK, Hellani A, Al-Mahed M, Al-Sheikh I. Spectrum of factor VIII mutations in Arab patients with severe haemophilia A. Haemophilia. 2008;14(3):484-488.
doi pubmed - Viel KR, Machiah DK, Warren DM, Khachidze M, Buil A, Fernstrom K, Souto JC, et al. A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and associations with plasma FVIII activity levels. Blood. 2007;109(9):3713-3724.
doi pubmed - Vidal F, Farssac E, Altisent C, Puig L, Gallardo D. Rapid hemophilia A molecular diagnosis by a simple DNA sequencing procedure: identification of 14 novel mutations. Thromb Haemost. 2001;85(4):580-583.
pubmed - Mahajan A, Chavali S, Kabra M, Chowdhury MR, Bharadwaj D. Molecular characterization of hemophilia B in North Indian families: identification of novel and recurrent molecular events in the factor IX gene. Haematologica. 2004;89(12):1498-1503.
pubmed - den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15(1):7-12.
doi - Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers. 2010;14(4):533-537.
doi pubmed - Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073-1081.
doi pubmed - Rodrigues JP, Levitt M, Chopra G. KoBaMIN: a knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res. 2012;40(Web Server issue):W323-328.
- Delarue M, Dumas P. On the use of low-frequency normal modes to enforce collective movements in refining macromolecular structural models. Proc Natl Acad Sci U S A. 2004;101(18):6957-6962.
doi pubmed - Darden T, Perera L, Li L, Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7(3):R55-60.
doi - Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277(5):1141-1152.
doi pubmed - Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548.
doi pubmed - Prlic A, Down TA, Kulesha E, Finn RD, Kahari A, Hubbard TJ. Integrating sequence and structural biology with DAS. BMC Bioinformatics. 2007;8:333.
doi pubmed - Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823-3835.
doi pubmed - Bromberg Y, Overton J, Vaisse C, Leibel RL, Rost B. In silico mutagenesis: a case study of the melanocortin 4 receptor. FASEB J. 2009;23(9):3059-3069.
doi pubmed - Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95-99.
doi - Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol. 2009;19(2):120-127.
doi pubmed - Kamal MZ, Ahmad S, Yedavalli P, Rao NM. Stability curves of laboratory evolved thermostable mutants of a Bacillus subtilis lipase. Biochim Biophys Acta. 2010;1804(9):1850-1856.
doi pubmed - Tavassoli K, Eigel A, Pollmann H, Horst J. Mutational analysis of ectopic factor VIII transcripts from hemophilia A patients: identification of cryptic splice site, exon skipping and novel point mutations. Hum Genet. 1997;100(5-6):508-511.
doi pubmed - Djambas Khayat C, Salem N, Chouery E, Corbani S, Moix I, Nicolas E, Morris MA, et al. Molecular analysis of F8 in Lebanese haemophilia A patients: novel mutations and phenotype-genotype correlation. Haemophilia. 2008;14(4):709-716.
doi pubmed - Schwaab R, Oldenburg J, Kemball-Cook G, Albert T, Juhler C, Hanfland P, Ingerslev J. Assay discrepancy in mild haemophilia A due to a factor VIII missense mutation (Asn694Ile) in a large Danish family. Br J Haematol. 2000;109(3):523-528.
doi pubmed - Awidi A, Ramahi M, Alhattab D, Mefleh R, Dweiri M, Bsoul N, Magablah A, et al. Study of mutations in Jordanian patients with haemophilia A: identification of five novel mutations. Haemophilia. 2010;16(1):136-142.
doi pubmed - Elmahmoudi H, Khodjet-el-khil H, Wigren E, Jlizi A, Zahra K, Pellechia D, Vinciguerra C, et al. First report of molecular diagnosis of Tunisian hemophiliacs A: identification of 8 novel causative mutations. Diagn Pathol. 2012;7:93.
doi pubmed - Citron M, Godmilow L, Ganguly T, Ganguly A. High throughput mutation screening of the factor VIII gene (F8C) in hemophilia A: 37 novel mutations and genotype-phenotype correlation. Hum Mutat. 2002;20(4):267-274.
doi pubmed - Waseem NH, Bagnall R, Green PM, Giannelli F. Start of UK confidential haemophilia A database: analysis of 142 patients by solid phase fluorescent chemical cleavage of mismatch. Haemophilia Centres. Thromb Haemost. 1999;81(6):900-905.
pubmed - Karimipoor M, Zeinali S, Nafissi N, Tuddenham EG, Lak M, Safaee R. Identification of factor IX mutations in Iranian haemophilia B patients by SSCP and sequencing. Thromb Res. 2007;120(1):135-139.
doi pubmed - Enayat MS, Karimi M, Chana G, Farjadian S, Theophilus BD, Hill FG. Mutation analysis in F9 gene of 17 families with haemophilia B from Iran. Haemophilia. 2004;10(6):751-755.
doi - Poort SR, Briet E, Bertina RM, Reitsma PH. A Dutch pedigree with mild hemophilia B with a missense mutation in the first EGF domain (factor IXOud en Nieuw Gastel). Nucleic Acids Res. 1989;17(14):5869.
doi pubmed - Jayandharan G, Shaji RV, Chandy M, Srivastava A. Identification of factor IX gene defects using a multiplex PCR and CSGE strategy-a first report. J Thromb Haemost. 2003;1(9):2051-2054.
doi pubmed - Tartary M, Vidaud D, Piao Y, Costa JM, Bahnak BR, Fressinaud E, Congard B, et al. Detection of a molecular defect in 40 of 44 patients with haemophilia B by PCR and denaturing gradient gel electrophoresis. Br J Haematol. 1993;84(4):662-669.
doi pubmed - Elmahmoudi H, Khodjet-El-Khil H, Ben-Amor M, Jlizi A, Zahra K, Meddeb B, Ben-Ammar-El-Gaaied A, et al. Identification of novel and recurrent mutations in Tunisian haemophilia B patients. Haemophilia. 2011;17(3):544-545.
doi pubmed - Fraser BM, Poon MC, Hoar DI. Identification of factor IX mutations in haemophilia B: application of polymerase chain reaction and single strand conformation analysis. Hum Genet. 1992;88(4):426-430.
doi pubmed - Montejo JM, Magallon M, Tizzano E, Solera J. Identification of twenty-one new mutations in the factor IX gene by SSCP analysis. Hum Mutat. 1999;13(2):160-165.
doi - Torkamani A, Verkhivker G, Schork NJ. Cancer driver mutations in protein kinase genes. Cancer Lett. 2009;281(2):117-127.
doi pubmed - Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17(4):263-270.
doi pubmed - Shen BW, Spiegel PC, Chang CH, Huh JW, Lee JS, Kim J, Kim YH, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111(3):1240-1247.
doi pubmed - Markoff A, Gerke V, Bogdanova N. Combined homology modelling and evolutionary significance evaluation of missense mutations in blood clotting factor VIII to highlight aspects of structure and function. Haemophilia. 2009;15(4):932-941.
doi pubmed - Higuchi M, Kazazian HH, Jr., Kasch L, Warren TC, McGinniss MJ, Phillips JA, 3rd, Kasper C, et al. Molecular characterization of severe hemophilia A suggests that about half the mutations are not within the coding regions and splice junctions of the factor VIII gene. Proc Natl Acad Sci U S A. 1991;88(16):7405-7409.
doi pubmed - Saunders RE, Perkins SJ. CoagMDB: a database analysis of missense mutations within four conserved domains in five vitamin K-dependent coagulation serine proteases using a text-mining tool. Hum Mutat. 2008;29(3):333-344.
doi pubmed - Gregory SG, Barlow KF, McLay KE, Kaul R, Swarbreck D, Dunham A, Scott CE, et al. The DNA sequence and biological annotation of human chromosome 1. Nature. 2006;441(7091):315-321.
doi pubmed - El-Maarri O, Herbiniaux U, Graw J, Schroder J, Terzic A, Watzka M, Brackmann HH, et al. Analysis of mRNA in hemophilia A patients with undetectable mutations reveals normal splicing in the factor VIII gene. J Thromb Haemost. 2005;3(2):332-339.
doi pubmed - Laurie AD, Smith MP. Effect of the F8 mutation c.1538-2A>T on pre-mRNA splicing. Haemophilia. 2009;15(6):1348-1350.
doi pubmed - Islam SI, Quadri MI. Spectrum of hereditary coagulation factor deficiencies in eastern province, Saudi Arabia. East Mediterr Health J. 1999;5(6):1188-1195.
pubmed - Yee TT, Williams MD, Hill FG, Lee CA, Pasi KJ. Absence of inhibitors in previously untreated patients with severe haemophilia A after exposure to a single intermediate purity factor VIII product. Thromb Haemost. 1997;78(3):1027-1029.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.