










| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Short Communication
Volume 8, Number 8, August 2016, pages 605-609
Can Multiple Hereditary Exostoses Overlap With Mesomelic Dysplasia?
Ali Al Kaissia, b, f, Maher Ben Ghachemc, Farid Ben Chehidad, Jochen G. Hofstaettera, b, Franz Grillb, Rudolf Gangerb, Susanne Gerit Kirchere
aLudwig Boltzmann Institute of Osteology at the Hanusch Hospital of WGKK and AUVA Trauma Centre Meidling, First Medical Department, Hanusch Hospital, Vienna, Austria
bOrthopaedic Hospital of Speising, Paediatric Department, Vienna, Austria
cDepartment of Paediatric Orthopaedics, Hopital d’infants, Tunis, Tunisia
dDepartment of Paediatric Radiology, Ibn Zohr Centre of Radiology, Tunis, Tunisia
eInstitute of Medical Chemistry, Medical University of Vienna, Austria
fCorresponding Author: Ali Al Kaissi, Ludwig Boltzmann Institute of Osteology at the Hanusch Hospital of WGKK and AUVA Trauma Center Meidling, First Medical Department, Hanusch Hospital and Orthopaedic Hospital of Speising Vienna, Austria
Manuscript accepted for publication May 25, 2016
Short title: Multiple Hereditary Exostoses
doi: http://dx.doi.org/10.14740/jocmr2593w
| Abstract | ▴Top |
Background: We studied an unusual combination of severe short stature, mesomelia (Leri-Weill dyschondrosteosis syndrome), and multiple exostosis in several family subjects over three generations. The pattern of inheritance was compatible with autosomal dominant.
Methods: Of 21 affected members over three generations, shortness of stature, associated with mesomelia resembling Leri-Weill dyschondrosteosis syndrome with no exostoses was evident in three family subjects. The rest of the family subjects manifested with normal height, and yet multiple exostoses. In this family, the skeletal manifestations were sufficiently variable for the presentation to be with either short stature or scoliosis, a Madelung’ deformity, or with severe hallux valgus associated with exostosis and with Leri-Weill dyschondrosteosis syndrome.
Results: Subjects with structural chromosomal aberrations of the proband IV-7, who manifested with normal height but with multiple exostoses were excluded via 20 CAG-banded mitoses (there were no microdeletions or microduplication after performing Array-CGH-analysis). In addition, DNA examination for subject IV-8 (male cousin of the proband showed short stature and Leri-Weill dyschondrosteosis syndrome) revealed no evidence of SHOX deletions.
Conclusion: We described a multigenerational non-consanguineous North African family , in which mesomelic dysplasia, whose clinical and radiological phenotypes resembled dyschondrosteosis, was a prominent feature in three family subjects. Multiple exostoses were evident in several other family subjects (most were with normal height). We would like to emphasize the variability in the phenotypic expression of multiple exostosis, especially the confusion that might arise when the condition appears both clinically and radiologically to be more complicated, and the overall picture might then be overlapped with one of the other bone dysplasias such as Leri-Weill dyschondrosteosis syndrome.
Keywords: Exostoses; Mesomelia; Array-CGH-analysis; No SHOX deletions
| Introduction | ▴Top |
Dyschondrosteosis is the most common variety of mesomelic short stature, affecting the middle segment of the extremities. It is characterized by forearms that are short and broad (the latter due to unusual curvature of the radius) and a Madelung deformity at the wrist. Genu valgum is common and the stature is reduced to between 137 and 152 cm in adults. Radiographs reveal bowing of the radius and ulna, whose distal ends form a “V” configuration, between which the carpals are wedged. The ulnar styloid is usually posteriorly dislocated. In the lower limbs, the tibia and fibula are short (with the latter being more severely affected) [1-3]. Exostoses are occasional, but not a frequent feature. On the other hand, autosomal dominant conditions of exostoses of the long bones, pelvis and scapula occur bilaterally. They occur at the periphery of the epiphyseal growth plate and the metaphyses. The exostoses can cause deformities such as bowing of the forearms, restriction of pronation and supination, tibia valga, obliquity of the distal tibial metaphysis and shortening of the fibula relative to the tibia [4]. Hip abnormalities, including coxa valga, are common and secondary to exostoses around the femoral heads [5]. Madelung’s deformity has been described in the literature as being not so disturbing to the patients and yet the interventions are so limited; on the other hand, exostosis has been described in certain instances, as being an occasional finding in such patients [6].
| Clinical Reports | ▴Top |
The proband IV-7, a 14-year-old female, was referred to the Department of Paediatric Orthopaedics because of bony lumps in her lower limbs and forearms. The child was born in Tunisia, to unrelated parents. At birth, her weight, length and head circumference were all around the 10th percentile. The mother was a 39-year-old gravida 4, abortus 0 woman, married to a 47-year-old unrelated man. The child’s first years of life were normal, but from the age of 10 years, the parents observed the presence of bony lumps over her limbs, though this was not a surprise for them, since her father (III-2), her 17-year-old male sib IV-8, and many relatives had similar lesions (Fig. 1).
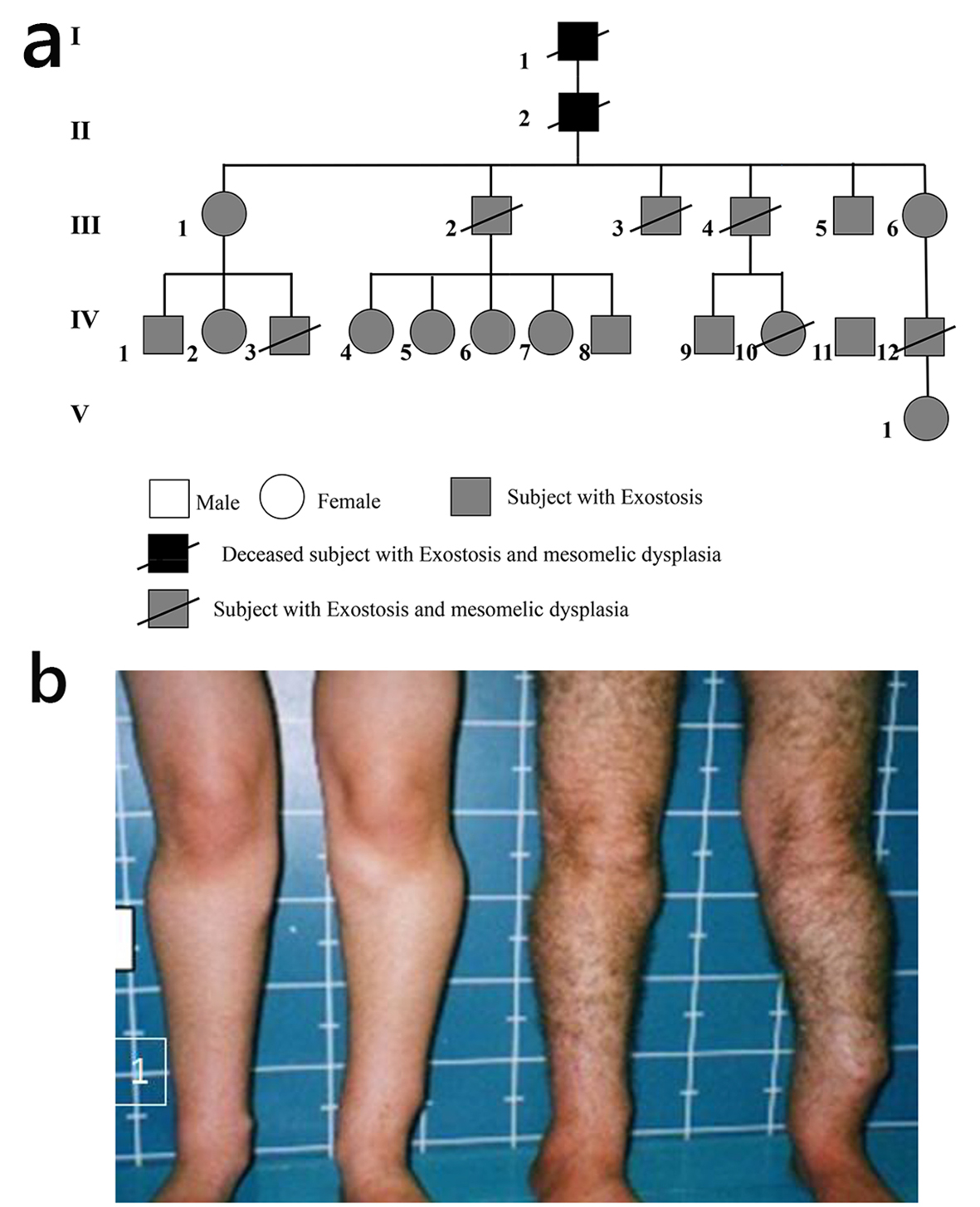 Click for large image | Figure 1. (a) Family pedigree. (b) The proband IV-7 and her father III-2 showed the multiple exostosis of the proband and of the father. |
On examination, the proband’s height was 155 cm. Her face was unremarkable except a degree of exophthalmos and a high palatine vault. Her joints were rigid, particularly over the wrists, hips, knees and the ankle joints, though the elbows were less stiff. There was an apparent dislocation of the inferior ulnar joint giving the impression of a Madelung deformity. There was marked stiffness over the cervical region secondary to the development of vertebral exostoses. Multiple exostoses were widely spread over the proximal and distal tibae, as well as over the proximal and the distal ulna. The upper thorax was narrow, and there was a thoracic scoliosis. Interestingly, subjects III-2 and III-4 (siblings) showed the severe short stature (-4 SD), short-limbed dwarfism dysplasia, but with no exostosis of subject III-4, while the next subject IV-8 (sibling) showed multiple exostosis, scoliosis but no mesomelia. Multiple scars of previous operations to remove exostosis along the scapula and the upper posterior left humerus were noted (Fig. 2). Structural chromosomal aberrations of the proband IV-7, who manifested with normal height but with multiple exostoses, were excluded via 20 CAG-banded mitoses (there were no microdeletions or microduplication after performing array-CGH-analysis). In addition, DNA examination for subject IV-8 (male cousin of the proband showed short stature and Leri-Weill dyschondrosteosis syndrome) revealed no evidence of SHOX deletions.
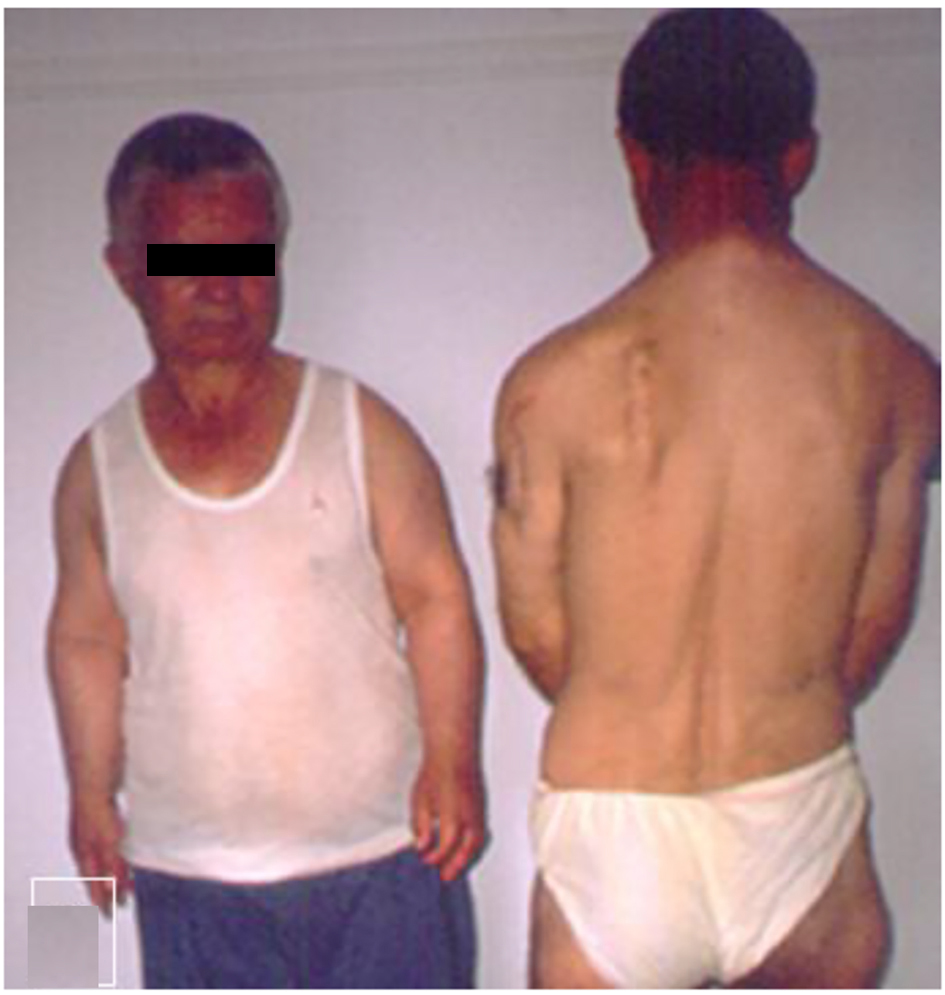 Click for large image | Figure 2. Subjects III-2 and III-4 (are siblings) showed the severe short stature (-4 SD), short-limbed dwarfism dysplasia but with no exostosis of subject III-4, next subject (sibling), whereas subject III-4 showed normal height, multiple exostosis, scoliosis but no mesomelia. Note multiple scars of previous operations to remove exostosis along the scapula and the upper posterior left humerus. |
Radiological examination
AP pelvis radiograph of the proband showed the dysplastic epiphyses bilaterally and the broadening of the metaphyses secondary bilateral and symmetrical chondromatous changes (Fig. 3a). X-ray of the forearm of the proband showed the multiple exostosis, bowing of the radius and ulna and the Madelung like deformity (Fig. 3b). X-ray cervical spines of the proband showed the involvement of the cervical spines characterized with cauwliflower-like mass of exostosis along C2 (Fig. 3c).
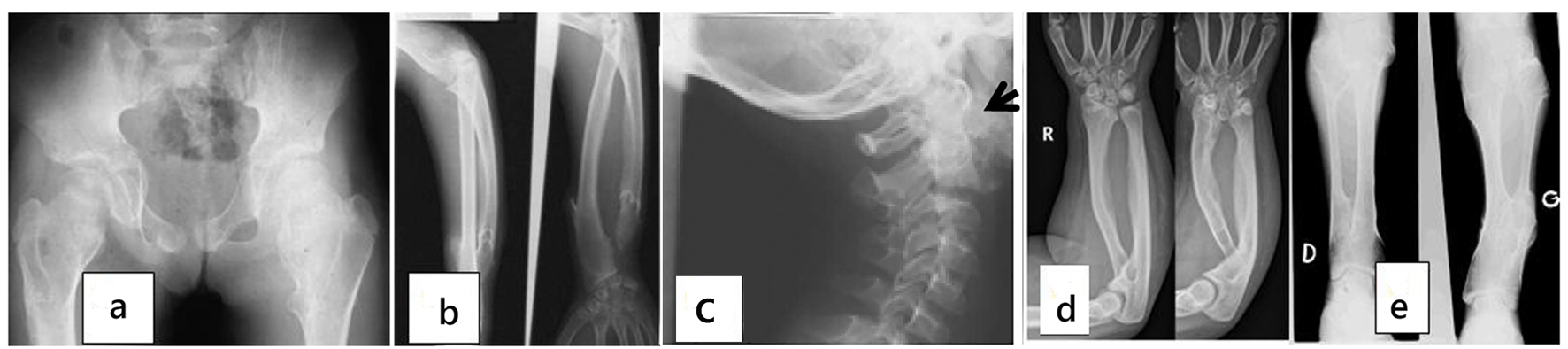 Click for large image | Figure 3. (a) AP pelvis radiograph of the proband showed the dysplastic epiphyses bilaterally and the broadening of the metaphyses secondary bilateral and symmetrical chondromatous changes. (b) X-ray of the forearm of the proband showed the multiple exostosis, bowing of the radius and ulna and the Madelung like deformity. (c) X-ray cervical spines of the proband showed the involvement of the cervical spines characterized with cauwliflower-like mass of exostosis along C2 (arrow). (d) AP radiograph of ulna of subject IV-12’s radius was short in relation to ulna. Ulnar and palmar slant of the radial articular surface and triangulation of the distal radial epiphyses were shown. The overall features were compatible with Leri-Weill dyschondrosteosis syndrome. (e) X-ray of the tibia and fibula of the father (III-2) showed the apparent shortening of the long bones and the multiple exostosis. |
AP radiograph showed ulna of subject IV-12’s radius was short in relation to ulna. Ulnar and palmar slant of the radial articular surface and triangulation of the distal radial epiphyses were noted. The overall features were compatible with Leri-Weill dyschondrosteosis syndrome (Fig. 3d). X-ray of the tibia and fibula of the father (III-2) showed the apparent shortening of the long bones and the multiple exostosis (Fig. 3e).
Other family members
Fourteen subjects were examined in detail out of the 21 by X-ray. Two subjects, subject III-4 and his nephew (VI-1), had the typical phenotype of a mesomelic dysplasia which clinically could easily be diagnosed for a severe dyschondrosteosis. Subject III-1 and her niece subject IV-10 both manifested with radiological signs of the disorder and multiple exostoses, whose presentation was more typical. The tallest subject in the family, subject IV-9, presented with a severe thoraco-lumbar scoliosis. On further examination, this was associated with multiple exostoses. As shown in Figure 2, it was extremely unusual to encounter the co-occurrence of the phenotype of mesomelia in subject III-4 and that the male sibling III-2 manifested with different clinical and radiographic phenotypes of normal height but with multiple exostoses.
| Discussion | ▴Top |
Dyschondrosteosis is the commonest variety of mesomelic dysplasia affecting only the middle segment of the extremities. Leri and Weil (1929) first identified this type and gave it the name dyschondrosteosis. It is not a rare disorder, it is of common occurrence in our department, and in vast majority presents with minimal disability; therefore, many remain undiagnosed [7].
It is characterized by moderate shortness of stature due to short tibia and fibula and short forearms, often with a wrist deformity, similar to Madelung’s deformity [8]. Anton et al [9] and Dannenberg et al [6] identified many cases said to be Madelung’s deformity but the presence of short stature and a hereditary influence suggested that they were instances of dyschondrosteosis.
Maroteaux and Lamy [1], Langer [2] and Dawe et al [10] have added further families and stress the clinical variation in the condition. Inheritance is autosomal dominant with only 50% penetrance.
Multiple exostosis is a dominantly inherited, genetically heterogeneous disorder. To date, three loci have been identified. Cook et al [11] found linkage to 8q24 markers in a subset of 11 families they studied. They estimated that 70% of families with multiple exostoses are linked to 8q24.
Wu et al [12] reported evidence for a second locus for multiple exostoses on 11p in two large families. Le Merrer et al [13] presented evidence for a third locus at 19p11-p13. 8q linked families are designated EXT1, 11p linked families are EXT2 and 19p families are EXT3.
Philippe et al [14] screened 17 families for mutations. Mutations were found in 12 (70%). Francannet et al [15] studied genotype/phenotype correlation in 42 French families. Sixty-four percent had an EXT1 mutation and 21% had EXT2 mutations. The most severe forms of the disease and malignant transformation of exostoses were associated with EXT1 mutations.
Pfeiffer [16] and Reinhardt and Pfeiffer [17] studied a kindred with 14 persons affected by hypoplasia of ulna and fibula in the pattern of a regular autosomal dominant. In addition to the shortening and characteristic dysplasia of the ulna and fibula, there were changes of the radius and tibia and of adjacent portions of the skeleton as well. This disorder could be allelic to dyschondrosteosis which is thought to represent the heterozygous state of Langer mesomelic dwarfism of the hypoplastic ulna, fibula and mandible type. Exostosis was not an accompanying deformity.
The 21 affected members in our family were ranging between short stature associated with mesomelia with Madelung’s deformity, and most were of normal height, and yet had the latter group the features of the multiple hereditary exostoses. In this family, the skeletal manifestations were sufficiently variable for the presentation to be with either short stature or scoliosis, a Madelung’ deformity, or with severe hallux valgus. A bowed and short ulna and radius (especially the ulna) have long been known to be part of multiple exostosis. Fairbank [18], Solomon [19], Bock and Reed [20] noted that the deformed and bent forearm bones could occur without evidence of exostosis. The radius is tethered to the ulna by the interoseus membrane and the short and bent ulna might cause radial bowing. Not all agree with Burgess and Cates [21], but in general, the ulnar carpal shift and the tethering result in a dislocated radial head.
There were in addition dysmorphic facial features in those who were affected. Their faces were long and they seemed to have sloping shoulders and a thin body habitus giving overall the appearance of tall stature, but the majority were indeed short. We report this family to emphasize how confusing the clinical and radiological features might be and neither the proband with multiple exostosis, nor the male cousin with mesomelia showed genotypic characterizations of the two disorders. The diagnosis and recognition of multiple exostoses might be straightforward in the majority of cases, but as in this family, the Madelung-type forearm deformity in association with mesomelic dysplasia and short limbed dwarfism did cause initial problems with the definite diagnosis. Patients with a Madelung-type deformity may have more complicated disease and multiple exostosis should be excluded as an underlying cause.
The overall clinico-radiographic phenotype in this family can be described as utterly unusual. The constellation of multiple hereditary exostosis, severe short stature, and mesomelic dysplasia in several patients over three generations can be described as a unique pattern of deformities. We would like to emphasize the variability in the phenotypic expression of multiple exostoses, especially the confusion that might arise when the condition appears both clinically and radiologically to be more complicated, and the overall picture might then be confused because of the overlap with Leri-Weill dyschondrosteosis syndrome.
Finally, we wish to stress that whether other genes on the same or a different chromosome affect expression and therefore clinical phenotype, or whether environmental effects are involved, is unclear and in order to approach to the final etiology to explain this unusual overlap, the necessity of further studies and research is mandatory.
Conflicts of Interest
Authors declare no conflicts of interest related to this manuscript.
| References | ▴Top |
- Maroteaux P, Lamy M. La dyschondrosteose. Sem Hop Paris. 1959;35:3467-3470.
- Langer LO, Jr. Mesomelic dwarfism of the hypoplastic ulna, fibula, mandible type. Radiology. 1967;89(4):654-660.
doi pubmed - Schiller S, Spranger S, Schechinger B, Fukami M, Merker S, Drop SL, Troger J, et al. Phenotypic variation and genetic heterogeneity in Leri-Weill syndrome. Eur J Hum Genet. 2000;8(1):54-62.
doi pubmed - Peterson HA. Deformities and problems of the forearm in children with multiple hereditary osteochondromata. J Pediatr Orthop. 1994;14(1):92-100.
doi pubmed - Porter DE, Benson MK, Hosney GA. The hip in hereditary multiple exostoses. J Bone Joint Surg Br. 2001;83(7):988-995.
doi pubmed - Dannenberg M, Anton JI, Spiegel MB. Madelung's deformity: consideration of its roentegenological diagnostic criteria. AJR. 1939;42:671-676.
- Leri A, Weill J. Une affection congenitale et symetrique du development osseux. La dyschondrosteose. Bull Soc Med Hop Paris. 1929;53:1491-1494.
- Herdman RC, Langer LO, Good RA. Dyschondrosteosis. The most common cause of Madelung's deformity. J Pediatr. 1966;68(3):432-441.
doi - Anton JI, Reitz GB, Spiegel MB. Madelung's Deformity. Ann Surg. 1938;108(3):411-439.
doi pubmed - Dawe C, Wynne-Davies R, Fulford GE. Clinical variation in dyschondrosteosis. A report on 13 individuals in 8 families. J Bone Joint Surg Br. 1982;64(3):377-381.
pubmed - Cook A, Raskind W, Blanton SH, Pauli RM, Gregg RG, Francomano CA, Puffenberger E, et al. Genetic heterogeneity in families with hereditary multiple exostoses. Am J Hum Genet. 1993;53(1):71-79.
pubmed - Wu YQ, Heutink P, de Vries BB, Sandkuijl LA, van den Ouweland AM, Niermeijer MF, Galjaard H, et al. Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11. Hum Mol Genet. 1994;3(1):167-171.
doi - Le Merrer M, Legeai-Mallet L, Jeannin PM, Horsthemke B, Schinzel A, Plauchu H, Toutain A, et al. A gene for hereditary multiple exostoses maps to chromosome 19p. Hum Mol Genet. 1994;3(5):717-722.
doi pubmed - Philippe C, Porter DE, Emerton ME, Wells DE, Simpson AH, Monaco AP. Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am J Hum Genet. 1997;61(3):520-528.
doi pubmed - Francannet C, Cohen-Tanugi A, Le Merrer M, Munnich A, Bonaventure J, Legeai-Mallet L. Genotype-phenotype correlation in hereditary multiple exostoses. J Med Genet. 2001;38(7):430-434.
doi pubmed - Pfeiffer RA. Beitrag zur erblichen Verkuerzung von Ulna und Fibula. In: Wiedemann, H-R. Dysostosen. Stuttgart: Gustav Fischer Verlag (pub.). 1966.
- Reinhardt K, Pfeiffer RA. Ulno-fibular dysplasia. An autosome-dominant hereditary micromesomelia resembling the Nievergelt syndrome. Fortschr Geb Rontgenstr Nuklearmed. 1967;107(3):379-391.
doi pubmed - Fairbank HA. Diaphysial aclasis; synonyms; multiple exostoses, hereditary deforming chondrodysplasia. J Bone and Joint Surg Br. 1949;31-B(1):105-113.
- Solomon L. Bone growth in diaphysial aclasis. J Bone Joint Surg Br. 1961;43-B:700-716.
pubmed - Bock GW, Reed MH. Forearm deformities in multiple cartilaginous exostoses. Skeletal Radiol. 1991;20(7):483-486.
doi pubmed - Burgess RC, Cates H. Deformities of the forearm in patients who have multiple cartilaginous exostosis. J Bone Joint Surg Am. 1993;75(1):13-18.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.